Structural analysis of nucleic acid
used to be a rather tedious process, especially for irregular, complicated RNA structures and nucleic acid-protein complexes (e.g., the large ribosomal subunit
1jj2/rr0033). Without valid base-pairing information as input, the various analysis software will produce meaningless results. The program
find_pair was originally created to solve this specific problem, by generating input file to
3DNA analysis routines (
analyze/cehs) directly from a PDB file.
In its core,
find_pair uses a pure geometric approach to identify
all possible pairs (Watson-Cricks or non-canonical pairs actually exist in a structure), their H-bonding patterns and helix context. Specifically, the
major criteria used are as follows:
- The distance between the origins of the two bases (as defined by their standard reference frames) must be less than certain limit (15.0 Å by default) - otherwise, they would be too far away to be called a pair.
- The vertical separation (i.e., stagger) between the two base planes must be less than certain limit (2.5 Å by default) - otherwise, they would be stacking instead of pairing.
- The angle between the two base z-axes (i.e., their normal vectors) is less than a cut-off (65.0° by default).
- There is at least one pair of nitrogen/oxygen base atoms that are within a H-bonding cut off distance (4.0 Å by default).
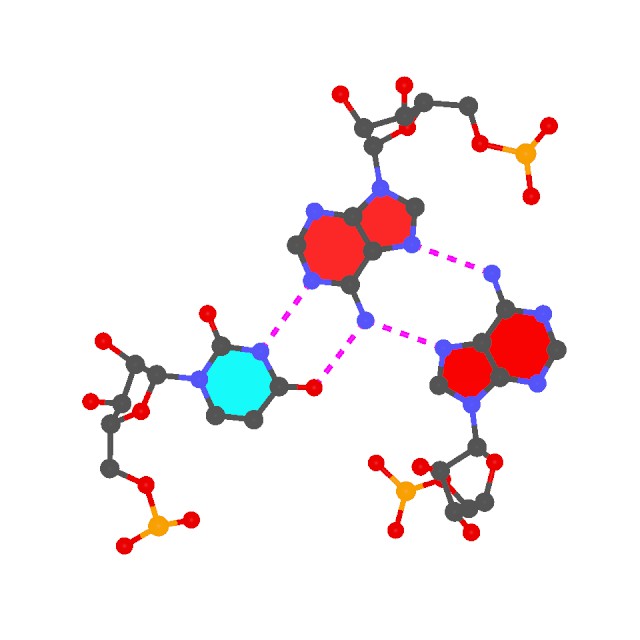
If two bases fulfill these geometric requirements, they are defined to be a pair, without taking consideration of their chemical constituents. Thus our method allows for identification of
unconventional pairs as easily as the canonical ones. The program then checks for
possible H-bonding patterns, whether the normal donor-acceptor (noted by '-' as in
O6 - N4 for a G·C pair) or the unusual donor-donor, acceptor-acceptor (noted by '*' as in
O2 * N3 for a C·C pair in
urx057). The non-canonical pairs, especially those with unusual H-bonding patterns, should be checked more carefully - they could be due to errors in structure determination, or they could have some special meaning/significance unnoticed previously.
The default criteria mentioned above are based on a survey of the NDB structures. Generally speaking, they are pretty generous and work quite well in the most common cases we've encountered. However, we are aware of the possibilities of special cases where some of them might be too restrict or too generous, thus leading to
find_pair to miss or produce superfluous base pairs. The default settings are stored in a text file named
misc_3dna.par under the directory
$X3DNA/config/ where users can modify as they see fit. Changes in that directory will have a global effect - wherever you run
find_pair on your system, the modified values will be used. Alternately, users could make a copy of
misc_3dna.par to their current working directory and change it over there for local effect. Note that the local setting has precedence over the global one.
As an example,
find_pair will miss the 127th base-pair
I:..53_:[.DT]T-----A[.DA]:.-53_:J in structure
1kx5/pd0287 in its default settings. This is because the H-bonding distance between
T:N3 - A:N1 is 4.20 Å and that for
T:O4 - A:N6 is 4.85 Å; both of them are larger than the default 4.0 Å cut off. Increasing the H-bonding criterion in file
misc_3dna.par from 4.0 Å to 5.0 Å will solve this problem. Please note that in 3DNA, users can start directly from an uncompressed PDB file, without having to extract the DNA fragment first:
- find_pair 1kx5.pdb 1kx5.inp to get input file for analyze
- analyze 1kx5.inp to get detailed structural parameters in file 1kx5.out
- The above two steps can be combined into one: find_pair 1kx5.pdb stdout | analyze stdin
In addition to (or instead of) manipulating parameters in
misc_3dna.par, oftentimes it may be preferable to manually edit
find_pair-generated base-piar files before feeding them into
analyze/cehs. This allows for maximum flexibility as to which pair to consider in calculating 3DNA structural parameters.
Also worth noting is the -p option of find_pair: without this option, find_pair locates base pairs in double-helical regions; thus the Watson-Crick pairs take precedence over the Wobble and other non-canonical pairs. With the -p, then all pairs and higher order base associations (i.e., triplets and above) are detected.



{kind=link}