Note added on 2020-05-12: mutate_bases is now obsoleted by DSSR 2.0.
See also:
The utility program
mutate_bases can be used to mutate bases in nucleic-acid-containing structures (DNA, RNA, and their complexes with ligands and proteins). It has two key and unique features: (1) the sugar-phosphate backbone conformation is untouched; (2) the base reference frame (position and orientation) is conserved,
i.e., the mutated structure shares the same base-pair/step parameters as those of the native structure.
The
mutate_bases program was created in response to repeated requests from 3DNA users over the years. Written as a standalone ANSI C program, it is on a par with other major 3DNA components (e.g.,
find_pair,
analyze,
rebuild and
fiber). The program was first released as a supplement to 3DNA v2.0, and then became an essential part of the v2.1 release.
Overall,
mutate_bases has been designed to solve the
in silico base mutation problem in a practical sense:
robust and efficient, getting its job done and then out of the way. The program can have many possible applications: in addition to perform base-pair mutations in DNA-protein complexes, it should also prove handy in RNA modeling and in providing initial structures for QM/MM/MD energy calculations, and in DNA/RNA modeling studies.
The standard command line help (
mutate_bases -h) is as below:
NAME
mutate_bases -- mutate bases, with backbone conformation unchanged
SYNOPSIS
mutate_bases [OPTIONS] mutinfo pdbfile outfile
DESCRIPTION
perform in silico base mutations of 3-dimensional nucleic acid
structures, with two key and unique features: (1) the sugar-
phosphate backbone conformation is untouched; (2) the base
reference frame (position and orientation) is reserved, i.e.,
the mutated structure shares the same base-pair/step
parameters as the original one.
-e enumeration of all bases in the structure
-l name of file, containing list of mutations
'mutinfo' can contain upto 5 fields for each mutation
[name=residue_name] [icode=insertion_code]
chain=chain_id seqnum=residue_number
mutation=residue_name
The five fields per mutation can be in any order or CaSe.
Each field can be abbreviated to its first character.
Multiple mutations specified per line are separated by ';'.
Fields in [] (i.e., name and icode) are optional.
Mutation info should be QUOTED to be taken as one entry.
INPUT
Nucleic-acid-containing structure file in PDB format
EXAMPLES
# mutate G2 in chain A of B-DNA 355d to Adenine
mutate_bases "c=a s=2 m=DA" 355d.pdb 355d_G2A.pdb
# mutate the second base-pair G-C to A-T in 355d
mutate_bases "c=a s=2 m=DA; c=B s=23 m=DT" 355d.pdb 355d_GC2AT.pdb
# the above also generates file 'mutations.dat'
# and the following command gives the same results
mutate_bases -l mutations.dat 355d.pdb 355d_GC2AT_v2.pdb
# mutate C74 in chain A of tRNA 1evv to U
mutate_bases "c=A s=74 m=U" 1evv.pdb 1evv_C74U.pdb
# list all bases to be tailored for mutation
mutate_bases -e 355d.pdb stdout
OUTPUT
mutated structure in PDB format, sharing the same backbone
conformation and base pair parameters as the original one.
SEE ALSO
analyze, find_pair, rebuild
AUTHOR
3DNA v2.1 (c) 2012 Dr. Xiang-Jun Lu (http://x3dna.org)
Now let's take advantage of the web to illustrate the key features of
mutate_bases using a set of worked examples. The scripts and corresponding data files & images are attached, so you can repeat the procedures in order to have a better understanding of how the program works.
In our
GpU dinucleotide platform paper, we reported a
previously unnoticed intra-dinucleotide sugar-phosphate H-bond that is unique to the GpU platform. This O2′(G)···O2P(U) H-bond readily rationalizes the over 60% occurrence of GpU over other platforms (e.g., ApA and UpC). Moreover, this H-bond has recently been
validated by state-of-the-art quantum-chemical techniques.
In this section, we will use
mutate_bases to answer the questions of (1) why GpU, not GpT? i.e., why the GpU platform is RNA-specific? (2) why no UpG platforms observed? i.e., why the GpU platform is directional? The
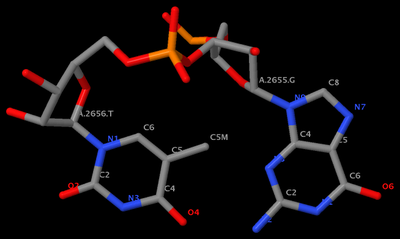
GpU platform (
1msy_gu.pdb) is derived from
PDB entry 1msy. The figure below shows the identity of the two nucleotides (G2655 and U2656 on chain A) and names of the base atoms.
You can run
find_pair and
analyze to the raw and mutated PDB files and verify that they indeed have the same base-pair parameters and backbone conformation.
To summarize, here is the command-script:
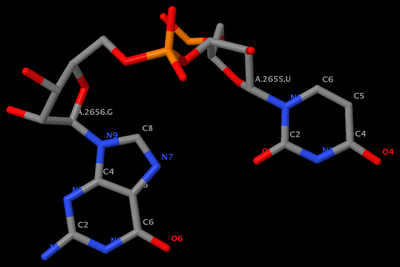
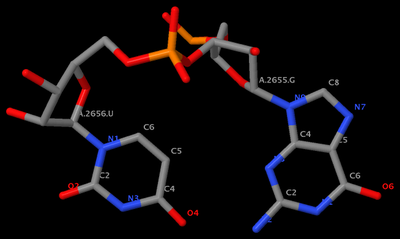
mutate_bases "c=a s=2656 m=t" 1msy_gu.pdb 1msy_gt.pdb
mutate_bases "c=a s=2655 m=u; c=a s=2656 m=g" 1msy_gu.pdb 1msy_ug.pdb
mutate_bases -l mutations.dat 1msy_gu.pdb 1msy_ug2.pdbThe PDB files referred:
Note all the images used in this post were generated using
Jmol. As much I like RasMol (v2.6.4), I am now gradually switching to Jmol and PyMOL.
Note added on Monday, July 17, 2017:
Single quotes in
mutate_bases command-line option have been replaced by double quotes so that the program also works in native Windows. See follow-up messages below.



 in helping me identify and fix this very subtle bug that can be traced back to v1.5! The tricky part is that it normally does not show up -- not on CentOS 5, Scientific Linux 6, Debian 5, Debian 6, Ubuntu 10.10 and OpenSuSE 11.3. Even if I compile 3DNA directly on Fedora 14 64bit, everything runs smoothly.
in helping me identify and fix this very subtle bug that can be traced back to v1.5! The tricky part is that it normally does not show up -- not on CentOS 5, Scientific Linux 6, Debian 5, Debian 6, Ubuntu 10.10 and OpenSuSE 11.3. Even if I compile 3DNA directly on Fedora 14 64bit, everything runs smoothly.