

1
Q&As related to x3DNA-DSSR; an online community for DNA/RNA structural bioinformatics x3DNA-DSSR · w3DNA
 Show Posts
Show Posts
This section allows you to view all posts made by this member. Note that you can only see posts made in areas you currently have access to.
Netiquette · Download · News · Gallery · G-quadruplexes · DSSR-Jmol · DSSR-PyMOL · Video Overview · DSSR v2.5.4 (DSSR Manual) · Homepage
2
Site announcements / On registration and posting
« on: April 19, 2025, 11:20:25 pm »
It has been 14 years since the Forum was created in 2011. Despite a four-year gap in NIH funding, we managed to keep the Forum operational. Maintaining and nurturing our community wasn't easy, but the users' enthusiasm in using and citing 3DNA/DSSR has kept us going. With the dedicated R24GM153869 grant, I am now committed to making the Forum even better.
Keeping the Forum spam-free is our top priority. In recent months, we have seen a dramatic increase in spams, which account for the majority of new registrations. That is why we have implemented 'Admin Approval' as the method of registration for new members. I carefully review each new registration to ensure only legitimate users are approved to join the Forum. Once approved, new users need to activate their accounts by clicking the activation link sent to their registered email address. I have noticed that some users did not activate their accounts upon approval. I normally send reminders to those inactivated users, but if they still do not respond in a few days, their registrations will be removed from the Forum.
It could also be the other way around: for example, the activation email sent from the 3DNA Forum might have been filtered out as spam by the user’s email agent. I have recently helped a few users with their registrations. If you have any questions or concerns about your registration, feel free to reach out to me directly via email. In today's age of AI, a personal touch goes a long way. Getting assistance directly from the developer ensures issues are resolved quickly and effectively.
I am dedicated to continuously enhancing X3DNA-DSSR, aiming to build it as a reputable brand symbolizing quality and value. Due to its exceptional functionality, ease of use, and direct support from the developer, X3DNA-DSSR significantly reduces the time and effort required compared to alternative solutions. Your comments, suggestions, and bug reports are greatly appreciated; I carefully consider every piece of user feedback, and always respond promptly. Specifically, I encourage you to openly share any challenges or negative experiences you encounter during installation or usage. Asking your questions on the public 3DNA Forum benefits not only yourself but also the wider user community.
Enclosed below is the Registration Agreement for the Forum
This forum is dedicated to topics generally related to the X3DNA-DSSR resource for the analysis, rebuilding, and visualization of 3D nucleic acid structures. To make the Forum a pleasant virtual community for all of us to learn from and contribute to, please be considerate and practice good netiquette (http://www.albion.com/netiquette/). See also the FAQ entry "How to make the best use of the Forum".
I strive to make the Forum spam free. Private emails (gmail.com, yahoo.com, qq.com, rambler.ru etc.) are not accepted; such registrations will be removed. Approved registrations that are not activated via email will be deleted. Activated accounts that are not accessed (logins) will be erased. Posts that are not 3DNA/DSSR related in the broad sense are taken as spams and are strictly forbidden. All administrative actions are performed without notification.
DSSR has completely superseded 3DNA (which is still maintained, but no new features other than bug fixes). DSSR integrates the disparate programs of 3DNA under one umbrella, and offers new advanced features, through a convenient interface. DSSR requires no set up or configuration: it just works. See the Overview Video and User Manual.
When posting on the Forum, please abide by the following rules:
0. Do your homework; read the FAQ and browse the Forum.
1. Ask your questions on the *public* 3DNA Forum instead of sending
xiangjun emails or personal messages. Additionally, please note
that your posts on the 3DNA Forum are in the *public domain*.
2. Be specific with your questions; provide a minimal, reproducible
example if possible; use attachments where appropriate.
3. Respond to requests for clarification. Failure to do so may result in
delay or no answer to your questions.
4. Summarize the solution to your problem from a user's perspective
by providing step-by-step details, for the community's benefit.
5+ Contribute back to the 3DNA project:
o Report bugs — including typos
o Make constructive suggestions — anything that can make 3DNA better
o Answer other users' questions
o Share your use cases in the "Users' contributions" section
In a nutshell, you are welcome to participate and should not hesitate to ask questions, but remember to play nice and preferably share what you learned! Please note that we do *not* tolerate spamming or off-topic trolling of any form.
Keeping the Forum spam-free is our top priority. In recent months, we have seen a dramatic increase in spams, which account for the majority of new registrations. That is why we have implemented 'Admin Approval' as the method of registration for new members. I carefully review each new registration to ensure only legitimate users are approved to join the Forum. Once approved, new users need to activate their accounts by clicking the activation link sent to their registered email address. I have noticed that some users did not activate their accounts upon approval. I normally send reminders to those inactivated users, but if they still do not respond in a few days, their registrations will be removed from the Forum.
It could also be the other way around: for example, the activation email sent from the 3DNA Forum might have been filtered out as spam by the user’s email agent. I have recently helped a few users with their registrations. If you have any questions or concerns about your registration, feel free to reach out to me directly via email. In today's age of AI, a personal touch goes a long way. Getting assistance directly from the developer ensures issues are resolved quickly and effectively.
I am dedicated to continuously enhancing X3DNA-DSSR, aiming to build it as a reputable brand symbolizing quality and value. Due to its exceptional functionality, ease of use, and direct support from the developer, X3DNA-DSSR significantly reduces the time and effort required compared to alternative solutions. Your comments, suggestions, and bug reports are greatly appreciated; I carefully consider every piece of user feedback, and always respond promptly. Specifically, I encourage you to openly share any challenges or negative experiences you encounter during installation or usage. Asking your questions on the public 3DNA Forum benefits not only yourself but also the wider user community.
Enclosed below is the Registration Agreement for the Forum
This forum is dedicated to topics generally related to the X3DNA-DSSR resource for the analysis, rebuilding, and visualization of 3D nucleic acid structures. To make the Forum a pleasant virtual community for all of us to learn from and contribute to, please be considerate and practice good netiquette (http://www.albion.com/netiquette/). See also the FAQ entry "How to make the best use of the Forum".
I strive to make the Forum spam free. Private emails (gmail.com, yahoo.com, qq.com, rambler.ru etc.) are not accepted; such registrations will be removed. Approved registrations that are not activated via email will be deleted. Activated accounts that are not accessed (logins) will be erased. Posts that are not 3DNA/DSSR related in the broad sense are taken as spams and are strictly forbidden. All administrative actions are performed without notification.
DSSR has completely superseded 3DNA (which is still maintained, but no new features other than bug fixes). DSSR integrates the disparate programs of 3DNA under one umbrella, and offers new advanced features, through a convenient interface. DSSR requires no set up or configuration: it just works. See the Overview Video and User Manual.
When posting on the Forum, please abide by the following rules:
0. Do your homework; read the FAQ and browse the Forum.
1. Ask your questions on the *public* 3DNA Forum instead of sending
xiangjun emails or personal messages. Additionally, please note
that your posts on the 3DNA Forum are in the *public domain*.
2. Be specific with your questions; provide a minimal, reproducible
example if possible; use attachments where appropriate.
3. Respond to requests for clarification. Failure to do so may result in
delay or no answer to your questions.
4. Summarize the solution to your problem from a user's perspective
by providing step-by-step details, for the community's benefit.
5+ Contribute back to the 3DNA project:
o Report bugs — including typos
o Make constructive suggestions — anything that can make 3DNA better
o Answer other users' questions
o Share your use cases in the "Users' contributions" section
In a nutshell, you are welcome to participate and should not hesitate to ask questions, but remember to play nice and preferably share what you learned! Please note that we do *not* tolerate spamming or off-topic trolling of any form.
3
Site announcements / DSSR-PyMOL enabled schematics on the covers of the RNA Journal
« on: December 16, 2024, 04:11:24 pm »Quote
Cover image provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).
See the 2020 paper titled "DSSR-enabled innovative schematics of 3D nucleic acid structures with PyMOL" in Nucleic Acids Research and the corresponding Supplemental PDF for details. Many thanks to Drs. Wilma Olson and Cathy Lawson for their help in the preparation of the illustrations.
June 2025 (link to the source)
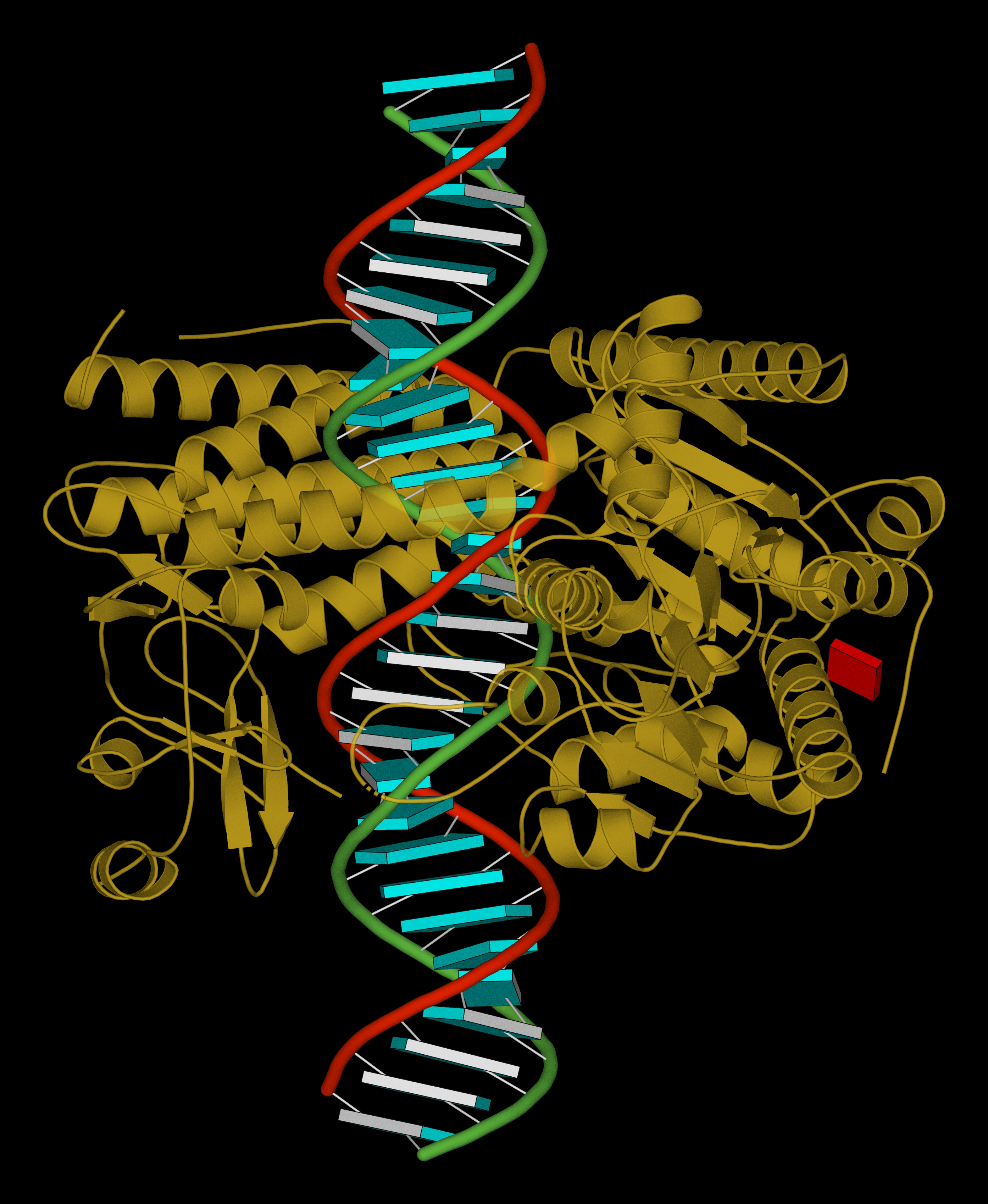
 | Structure of a group II intron ribonucleoprotein in the pre-ligation state (PDB id: 8T2R; Xu L, Liu T, Chung K, Pyle AM. 2023. Structural insights into intron catalysis and dynamics during splicing. Nature 624: 682–688). The pre-ligation complex of the Agathobacter rectalis group II intron reverse transcriptase/maturase with intron and 5′-exon RNAs makes it possible to construct a picture of the splicing active site. The intron is depicted by a green ribbon, with bases and Watson-Crick base pairs represented as color-coded blocks: A/A-U in red, C/C-G in yellow, G/G-C in green, U/U-A in cyan; the 5′-exon is shown by white spheres and the protein by a gold ribbon. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74). |
May 2025 (link to the source)
 | Complex of terminal uridylyltransferase 7 (TUT7) with pre-miRNA and Lin28A (PDB id: 8OPT; Yi G, Ye M, Carrique L, El-Sagheer A, Brown T, Norbury CJ, Zhang P, Gilbert RJ. 2024. Structural basis for activity switching in polymerases determining the fate of let-7 pre-miRNAs. Nat Struct Mol Biol 31: 1426–1438). The RNA-binding pluripotency factor LIN28A invades and melts the RNA and affects the mechanism of action of the TUT7 enzyme. The RNA backbone is depicted by a red ribbon, with bases and Watson-Crick base pairs represented as color-coded blocks: A/A-U in red, C/C-G in yellow, G/G-C in green, U/U-A in cyan; TUT7 is represented by a gold ribbon and LIN28A by a white ribbon. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74). |
April 2025 (link to the source)
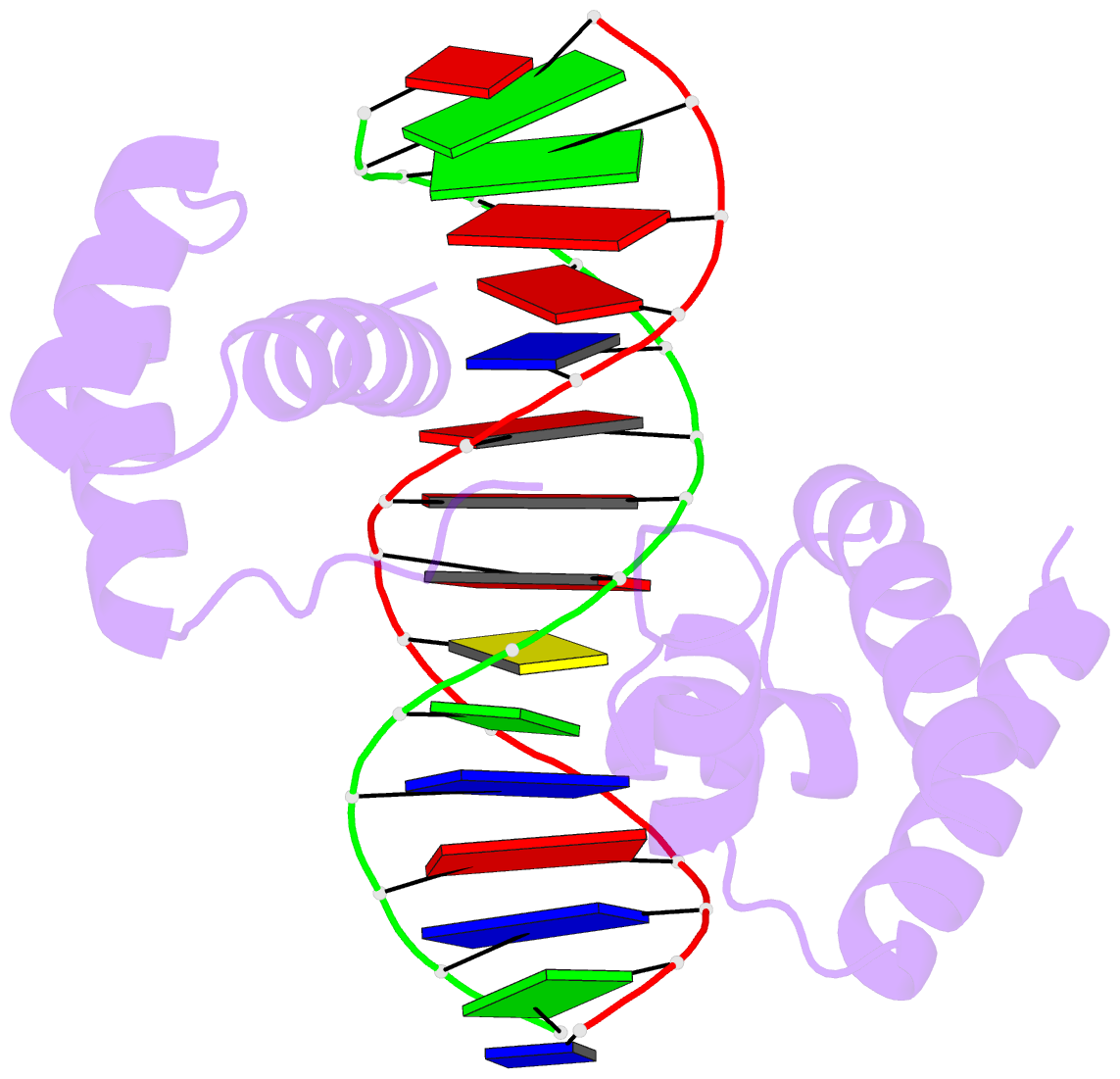
 | Cryo-EM structure of the pre-B complex (PDB id: 8QP8; Zhang Z, Kumar V, Dybkov O, Will CL, Zhong J, Ludwig SE, Urlaub H, Kastner B, Stark H, Lührmann R. 2024. Structural insights into the cross-exon to cross-intron spliceosome switch. Nature 630: 1012–1019). The pre-B complex is thought to be critical in the regulation of splicing reactions. Its structure suggests how the cross-exon and cross-intron spliceosome assembly pathways converge. The U4, U5, and U6 snRNA backbones are depicted respectively by blue, green, and red ribbons, with bases and Watson-Crick base pairs shown as color-coded blocks: A/A-U in red, C/C-G in yellow, G/G-C in green, U/U-A in cyan; the proteins are represented by gold ribbons. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74). |
February 2025 (link to the source)
 | Structure of the Hendra henipavirus (HeV) nucleoprotein (N) protein-RNA double-ring assembly (PDB id: 8C4H; Passchier TC, White JB, Maskell DP, Byrne MJ, Ranson NA, Edwards TA, Barr JN. 2024. The cryoEM structure of the Hendra henipavirus nucleoprotein reveals insights into paramyxoviral nucleocapsid architectures. Sci Rep 14: 14099). The HeV N protein adopts a bi-lobed fold, where the N- and C-terminal globular domains are bisected by an RNA binding cleft. Neighboring N proteins assemble laterally and completely encapsidate the viral genomic and antigenomic RNAs. The two RNAs are depicted by green and red ribbons. The U bases of the poly(U) model are shown as cyan blocks. Proteins are represented as semitransparent gold ribbons. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74). |
January 2025 (link to the source)
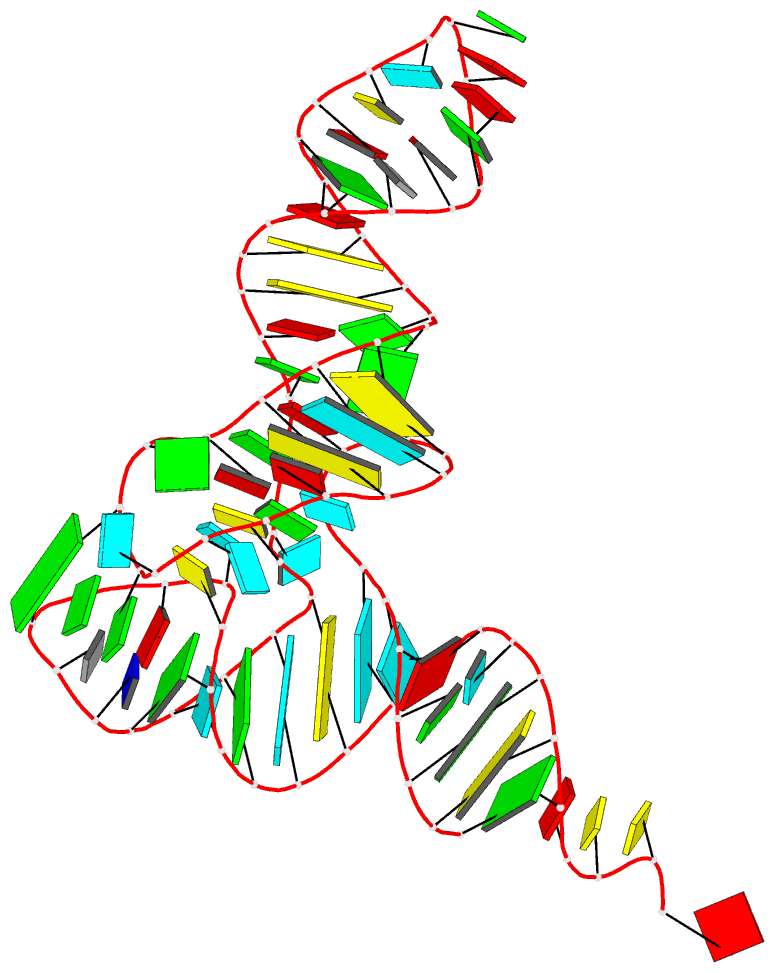
 | Structure of the helicase and C-terminal domains of Dicer-related helicase-1 (DRH-1) bound to dsRNA (PDB id: 8T5S; Consalvo CD, Aderounmu AM, Donelick HM, Aruscavage PJ, Eckert DM, Shen PS, Bass BL. 2024. Caenorhabditis elegans Dicer acts with the RIG-I-like helicase DRH-1 and RDE-4 to cleave dsRNA. eLife 13: RP93979). Cryo-EM structures of Dicer-1 in complex with DRH-1, RNAi deficient-4 (RDE-4), and dsRNA provide mechanistic insights into how these three proteins cooperate in antiviral defense. The dsRNA backbone is depicted by green and red ribbons. The U-A pairs of the poly(A)·poly(U) model are shown as long rectangular cyan blocks, with minor-groove edges colored white. The ADP ligand is represented by a red block and the protein by a gold ribbon. Cover image provided by X3DNA-DSSR, an NIGMS National Resource for structural bioinformatics of nucleic acids (R24GM153869; skmatics.x3dna.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74). |
How are the detailed steps used to generate the above cover image, using DSSR v2.4.6-2024nov15:
Code: Bash
- # Download the PDB structure in cif format from RCSB
- wget https://files.rcsb.org/download/8t5s-assembly1.cif -O 8t5s.cif
- # Run DSSR with the following settings. See below for file "8t5s-specific.pml"
- x3dna-dssr -i=8t5s.cif \
- --blocview=png-session-black \
- --block-file=wc-g4 \
- --block-depth=1.0 \
- --pymol-ray-size=5000 \
- --pymol-aa-color=gold \
- --block_color="minor:white" \
- --cartoon-cmd-file=8t5s-specific.pml \
- -o=8t5s.pml
- # Run PyMOL to render the image. Here it is from the command line.
- # You can also load the 8t5s.pml into PyMOL interactively.
- # Generate file "8t5s-pymol.png"
- pymol -Qkc 8t5s.pml
- # Use ImageMagick to trim extra margins
- magick 8t5s-pymol.png -trim +repage -bordercolor black -border 120 8t5s.png
Note the setting of black background, and coloring of minor-groove edge in white. By default, DSSR-PyMOL renders with a white background and black minor-groove edges, as in 8t5s. Whenever feasible, I've integrated features into DSSR to automate routine tasks as much as possible.
Here are the related files:
- 8t5s-specific.pml -- specific settings
- 8t5s.pse -- PyMOL session file
- 8t5s.pml -- PyMOL script file
 -- ray-traced image
-- ray-traced image
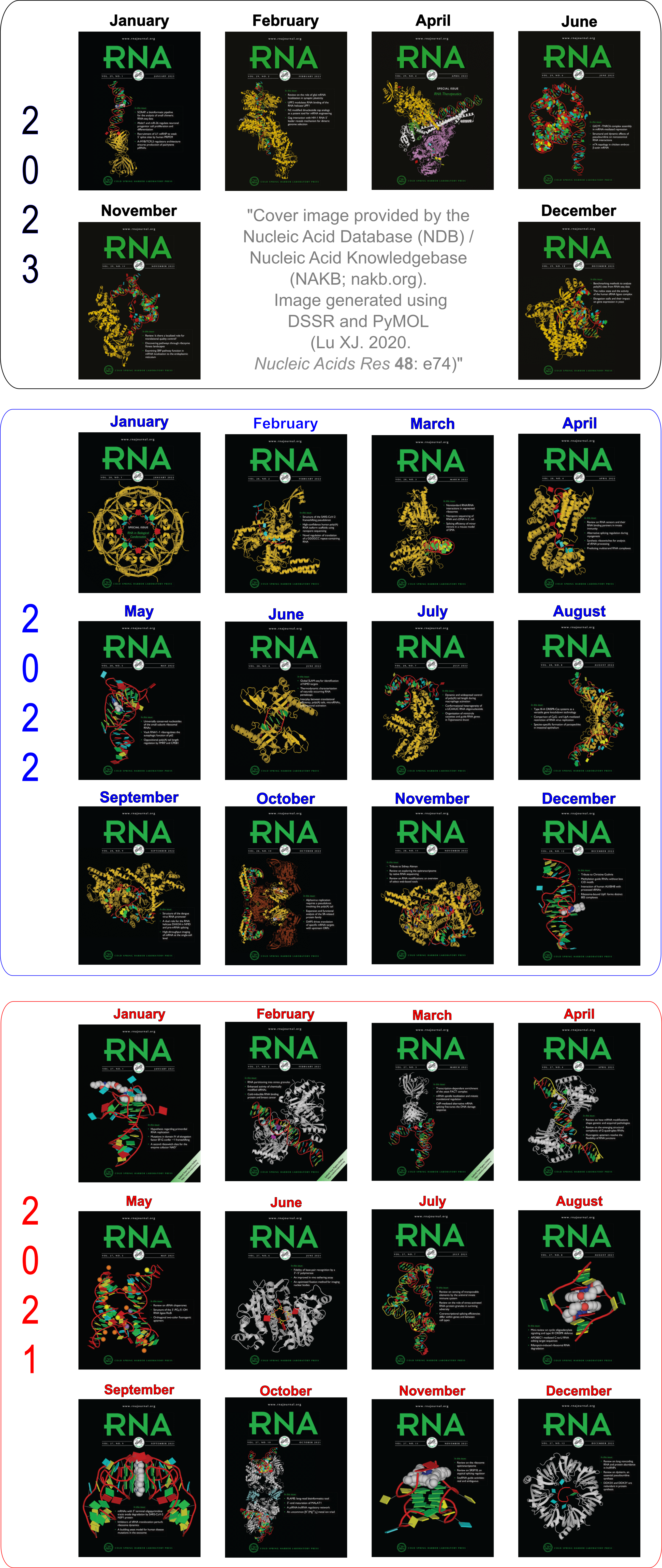
Moreover, the following 30 [12(2021) + 12(2022) + 6(2023)] cover images of the RNA Journal were generated by the NAKB (nakb.org).
Quote
Cover image provided by the Nucleic Acid Database (NDB)/Nucleic Acid Knowledgebase (NAKB; nakb.org). Image generated using DSSR and PyMOL (Lu XJ. 2020. Nucleic Acids Res 48: e74).
4
Site announcements / X3DNA-DSSR is funded and DSSR Basic is free for academic users
« on: September 25, 2024, 10:31:18 am »
Dear 3DNA/DSSR users,
It gives me great pleasure to announce that the 3DNA/DSSR project is now funded by the NIH R24GM153869 grant, "X3DNA-DSSR: a resource for structural bioinformatics of nucleic acids". I am deeply grateful for the opportunity to continue working on a project that has basically defined who I am. It was a tough time during the funding gap over the past few years. Nevertheless, I have experienced and learned a lot, and witnessed miracles enabled by enthusiastic users.
Since late 2020 when I lost my R01 grant, DSSR has been licensed by the Columbia Technology Ventures (CTV). I appreciate the numerous users (including big pharma) who purchased a DSSR Pro License or a DSSR Basic paid License. Thanks to the NIH R24GM153869 grant, we are pleased to provide DSSR Basic free of charge to the academic community. Academic Users may submit a license request for DSSR Basic or DSSR Pro by clicking "Express Licensing" on the CTV landing page. Commercial users may inquire about pricing and licensing terms by emailing techtransfer@columbia.edu, copying xiangjun@x3dna.org.
The current version of DSSR is v2.4.5-2024sep24 which contains miscellaneous bug fixes (e.g., chain id with > 4 chars) and minor improvements. This release synchronizes with the new R24 funding, which will bring the project to the next stage. All existing users are encouraged to upgrade their installation.
Lots of exciting things will happen for the project. The first important thing is to make DSSR freely accessible to the academic community. I'm now starting to monitor the Forum closely and answer users questions promptly.
I am committed to making DSSR a brand that stands for quality and value. By virtue of its unmatched functionality, usability, and support, DSSR saves users a substantial amount of time and effort when compared to other options. My track record throughout the years has unambiguously demonstrated my dedication to this solid software product.
Xiang-Jun
DSSR Basic contains all features described in the three DSSR-related papers, and include the originally separate SNAP program (still unpublished) for analyzing DNA/RNA-protein complexes. The Pro version integrates the classic 3DNA functionality, plus advanced modeling routines, with email/Zoom/phone support.
It gives me great pleasure to announce that the 3DNA/DSSR project is now funded by the NIH R24GM153869 grant, "X3DNA-DSSR: a resource for structural bioinformatics of nucleic acids". I am deeply grateful for the opportunity to continue working on a project that has basically defined who I am. It was a tough time during the funding gap over the past few years. Nevertheless, I have experienced and learned a lot, and witnessed miracles enabled by enthusiastic users.
Since late 2020 when I lost my R01 grant, DSSR has been licensed by the Columbia Technology Ventures (CTV). I appreciate the numerous users (including big pharma) who purchased a DSSR Pro License or a DSSR Basic paid License. Thanks to the NIH R24GM153869 grant, we are pleased to provide DSSR Basic free of charge to the academic community. Academic Users may submit a license request for DSSR Basic or DSSR Pro by clicking "Express Licensing" on the CTV landing page. Commercial users may inquire about pricing and licensing terms by emailing techtransfer@columbia.edu, copying xiangjun@x3dna.org.
The current version of DSSR is v2.4.5-2024sep24 which contains miscellaneous bug fixes (e.g., chain id with > 4 chars) and minor improvements. This release synchronizes with the new R24 funding, which will bring the project to the next stage. All existing users are encouraged to upgrade their installation.
Lots of exciting things will happen for the project. The first important thing is to make DSSR freely accessible to the academic community. I'm now starting to monitor the Forum closely and answer users questions promptly.
I am committed to making DSSR a brand that stands for quality and value. By virtue of its unmatched functionality, usability, and support, DSSR saves users a substantial amount of time and effort when compared to other options. My track record throughout the years has unambiguously demonstrated my dedication to this solid software product.
Xiang-Jun
DSSR Basic contains all features described in the three DSSR-related papers, and include the originally separate SNAP program (still unpublished) for analyzing DNA/RNA-protein complexes. The Pro version integrates the classic 3DNA functionality, plus advanced modeling routines, with email/Zoom/phone support.
6
Site announcements / DSSR-enabled innovative PyMOL schematics in the covers of the RNA Journal
« on: December 20, 2021, 11:31:10 am »
The DSSR-PyMOL schematics have been featured in all 12 cover images (January to December) of the RNA Journal in 2021. Moreover, the January 2022 issue of RNA continues to highlight DSSR-enabled schematics (see the note below). In the current Covid-19 pandemic, this cover seems to be a fit for the upcoming Christmas holiday season.
Thanks to Dr. Cathy Lawson at the NDB for generating these cover images using DSSR and PyMOL for the RNA Journal. I'm gratified that the 2020 NAR paper is explicitly acknowledged: it's the first time I've published as a single author in my scientific career.

Did you know that you can easily generate similar DSSR-PyMOL schematics via the http://skmatic.x3dna.org/ website? It is "simple and effective", "good for teaching", and has been highly recommended by Dr. Quentin Vicens (CU Denver) in FacultyOpinions.com.
The 12 PDB structures illustrated in the 2021 covers are:
Quote
Ebola virus matrix protein octameric ring (PDB id: 7K5L; Landeras-Bueno S, Wasserman H, Oliveira G, VanAernum ZL, Busch F, Salie ZL, Wysocki VH, Andersen K, Saphire EO. 2021. Cellular mRNA triggers structural transformation of Ebola virus matrix protein VP40 to its essential regulatory form. Cell Rep 35: 108986). The Ebola virus matrix protein (VP40) forms distinct structures linked to distinct functions in the virus life cycle. VP40 forms an octameric ring-shaped (D4 symmetry) assembly upon binding of RNA and is associated with transcriptional control. RNA backbone is displayed as a red ribbon; block bases use NDB colors: A—red, G—green, U—cyan; protein is displayed as a gold ribbon. Cover image provided by the Nucleic Acid Database (ndbserver.rutgers.edu). Image generated using DSSR and PyMOL (Lu XJ. 2020. _Nucleic Acids Res_ *48*: e74).
Thanks to Dr. Cathy Lawson at the NDB for generating these cover images using DSSR and PyMOL for the RNA Journal. I'm gratified that the 2020 NAR paper is explicitly acknowledged: it's the first time I've published as a single author in my scientific career.
Did you know that you can easily generate similar DSSR-PyMOL schematics via the http://skmatic.x3dna.org/ website? It is "simple and effective", "good for teaching", and has been highly recommended by Dr. Quentin Vicens (CU Denver) in FacultyOpinions.com.
The 12 PDB structures illustrated in the 2021 covers are:
- January 2021 "iMango-III fluorescent aptamer (PDB id: 6PQ7; Trachman III RJ, Stagno JR, Conrad C, Jones CP, Fischer P, Meents A, Wang YX, Ferre-D'Amare AR. 2019. Co-crystal structure of the iMango-III fluorescent RNA aptamer using an X-ray free-electron laser. Acta Cryst F 75: 547). Upon binding TO1-biotin, the iMango-III aptamer achieves the largest fluorescence enhancement reported for turn-on aptamers (over 5000-fold)."
- February 2021 "Human adenosine deaminase (E488Q mutant) acting on dsRNA (PDB id: 6VFF; Thuy-Boun AS, Thomas JM, Grajo HL, Palumbo CM, Park S, Nguyen LT, Fisher AJ, Beal PA. 2020. Asymmetric dimerization of adenosine deaminase acting on RNA facilitates substrate recognition. Nucleic Acids Res. https://doi.org/10.1093/nar/gkaa532). Adenosine deaminase enzymes convert adenosine to inosine in duplex RNA, a modification that strongly affects RNA structure and function in multiple ways."
- March 2021 "Hepatitis A virus IRES domain V in complex with Fab (PDB id: 6MWN; Koirala D, Shao Y, Koldobskaya Y, Fuller JR, Watkins AM, Shelke SA, Pilipenko EV, Das R, Rice PA, Piccirilli JA. 2019. A conserved RNA structural motif for organizing topology within picornaviral internal ribosome entry sites. Nat Commun 10: 3629)."
- April 2021 "Mouse endonuclease V in complex with 23mer RNA (PDB id: 6OZO; Wu J, Samara NL, Kuraoka I, Yang W. 2019. Evolution of inosine-specific endonuclease V from bacterial DNase to eukaryotic RNase. Mol Cell 76: 44). Endonuclease V cleaves the second phosphodiester bond 3′ to a deaminated adenosine (inosine). Although highly conserved, EndoV change substrate preference from DNA in bacteria to RNA in eukaryotes."
- May 2021 "Manganese riboswitch from Xanthmonas oryzae (PDB id: 6N2V; Suddala KC, Price IR, Dandpat SS, Janeček M, Kührová P, Šponer J, Banáš P, Ke A, Walter NG. 2019. Local-to-global signal transduction at the core of a Mn2+ sensing riboswitch. Nat Commun 10: 4304). Bacterial manganese riboswitches control the expression of Mn2+ homeostasis genes. Using FRET, it was shown that an extended 4-way-junction samples transient docked states in the presence of Mg2+ but can only dock stably upon addition of submillimolar Mn2+."
- June 2021 "Sulfolobus islandicus Csx1 RNase in complex with cyclic RNA activator (PDB id: 6R9R; Molina R, Stella S, Feng M, Sofos N, Jauniskis V, Pozdnyakova I, Lopez-Mendez B, She Q, Montoya G. 2019. Structure of Csx1-cOA4 complex reveals the basis of RNA decay in Type III-B CRISPR-Cas. Nat Commun 10: 4302). CRISPR-Cas multisubunit complexes cleave ssRNA and ssDNA, promoting the generation of cyclic oligoadenylate (cOA), which activates associated CRISPR-Cas RNases. The Csx1 RNase dimer is shown with cyclic (A4) RNA bound."
- July 2021 "M. tuberculosis ileS T-box riboswitch in complex with tRNA (PDB id: 6UFG; Battaglia RA, Grigg JC, Ke A. 2019. Structural basis for tRNA decoding and aminoacylation sensing by T-box riboregulators. Nat Struct Mol Biol 26: 1106). T-box riboregulators are a class of cis-regulatory RNAs that govern the bacterial response to amino acid starvation by binding, decoding, and reading the aminoacylation status of specific transfer RNAs."
- August 2021 "CAG repeats recognized by cyclic mismatch binding ligand (PDB id: 6QIV; Mukherjee S, Blaszczyk L, Rypniewski W, Falschlunger C, Micura R, Murata A, Dohno C, Nakatan K, Kiliszek A. 2019. Structural insights into synthetic ligands targeting A–A pairs in disease-related CAG RNA repeats. Nucleic Acids Res 47:10906). A large number of hereditary neurodegenerative human diseases are associated with abnormal expansion of repeated sequences. RNA containing CAG repeats can be recognized by synthetic cyclic mismatch-binding ligands such as the structure shown."
- September 2021 "Corn aptamer complex with fluorophore Thioflavin T (PDB id: 6E81; Sjekloca L, Ferre-D'Amare AR. 2019. Binding between G quadruplexes at the homodimer interface of the Corn RNA aptamer strongly activates Thioflavin T fluorescence. Cell Chem Biol 26: 1159). The fluorescent compound Thioflavin T, widely used for the detection of amyloids, is bound at the dimer interface of the homodimeric G-quadruplex-containing RNA Corn aptamer."
- October 2021 "Cas9 nuclease-sgRNA complex with anti-CRISPR protein inhibitor (PDB id: 6JE9; Sun W, Yang J, Cheng Z, Amrani N, Liu C, Wang K, Ibraheim R, Edraki A, Huang X, Wang M, et al. 2019. Structures of Neisseria meningitidis Cas9 complexes in catalytically poised and anti-CRISPR-inhibited states. Mol Cell 76: 938–952.e5). Nme1Cas9, a compact nuclease for in vivo genome editing. AcrIIC3 is an anti-CRISPR protein inhibitor."
- November 2021 "Two-quartet RNA parallel G-quadruplex complexed with porphyrin (PDB id: 6JJI; Zhang Y, Omari KE, Duman R, Liu S, Haider S, Wagner A, Parkinson GN, Wei D. 2020. Native de novo structural determinations of non-canonical nucleic acid motifs by X-ray crystallography at long wavelengths. Nucleic Acids Res 48: 9886–9898)."
- December 2021 "Structure of S. pombe Lsm1–7 with RNA, polyuridine with 3' guanosine (PDB id: 6PPV; Montemayor EJ, Virta JM, Hayes SM, Nomura Y, Brow DA, Butcher SE. 2020. Molecular basis for the distinct cellular functions of the Lsm1–7 and Lsm2–8 complexes. RNA 26: 1400–1413). Eukaryotes possess eight highly conserved Lsm (like Sm) proteins that assemble into circular, heteroheptameric complexes, bind RNA, and direct a diverse range of biological processes. Among the many essential functions of Lsm proteins, the cytoplasmic Lsm1–7 complex initiates mRNA decay, while the nuclear Lsm2–8 complex acts as a chaperone for U6 spliceosomal RNA."
7
Site announcements / BioExcel webinar on DSSR
« on: November 23, 2021, 11:38:53 am »
On December 9, 2021, at 15:00 CET, I will present a BioExcel webinar titled "X3DNA-DSSR, a resource for structural bioinformatics of nucleic acids."
For the record, the screenshot of the announcement is shown below:

- The announcement is available here: https://bioexcel.eu/webinar-x3dna-dssr-a-resource-for-structural-bioinformatics-of-nucleic-acids-2021-12-09/.
- Here is the registration URL: https://us02web.zoom.us/webinar/register/WN_g55wmd9ETmeQwQuLfjH57g.
For the record, the screenshot of the announcement is shown below:
8
Site announcements / No more grant funding for 3DNA/DSSR
« on: October 30, 2021, 09:58:15 pm »Quote from: Columbia Technology Ventures (CTV)
Due to a lack of governmental funding support, we are no longer able to provide DSSR free of charge to the community. Academic users may submit a license request for DSSR Basic or DSSR Pro by clicking "Express Licensing". Commercial users may inquire about pricing and licensing terms by emailing techtransfer@columbia.edu, copying xiangjun@x3dna.org.
DSSR Pro excels in structural bioinformatics of RNA, DNA, and their protein complexes. The software has completely superseded 3DNA, and is being continuously improved. Revenue from licensing supports the development and availability of DSSR.
I am committed to making DSSR a brand that stands for quality and value. By virtue of its unmatched functionality, usability, and support, DSSR saves users a substantial amount of time and effort when compared to other options.
I designed, implemented, documented, and have continuously improved and supported DSSR. As a result, DSSR users may expect a rapid and concrete answer to their questions. My track record throughout the years has unambiguously demonstrated my dedication to DSSR. I strive to ensure that paying users' trust in DSSR is well-founded by providing them with the best services possible.
As a general rule, the CTV does not provide an evaluation license of DSSR. Potential users should watch the DSSR overview video (20m), browse the Forum, and read DSSR-related papers. If they still have questions or want to see a live demo, I would be pleased to accommodate them. Although more DSSR licenses are definitely beneficial, I do not have the time or desire to directly promote the product, including sending bulk emails to registered users of the Forum. As the developer, I can only strive to make DSSR the best it can be and let the rest sort itself out. I am a strong believer of the old Chinese saying: "酒香不怕巷子深" (Good wine needs no bush).
3DNA is obsolete and no longer maintained or supported. Thanks to the revenue from DSSR licenses, however, the following web-based resources remain accessible to the general public:
- http://web.x3dna.org/ (Web 3DNA 2.0)
- http://skmatic.x3dna.org/ (DSSR-enabled block-view schematics)
- http://G4.x3dna.org/ (DSSR-annotated G-quadruplexes in the PDB)
9
Site announcements / Clarification on DSSR licensing
« on: May 31, 2021, 01:58:55 pm »
Once in a while I receive emails from prospective users, both commercial and academic, about DSSR licensing from the Columbia Technology Venture (CTV). Louis made the following explicit suggestion in a recent thread titled "Bug or feature (?) : residue numbering not understood":
There are two types of DSSR Pro licenses, as shown below:
The free DSSR Basic academic license is no longer available as of November 11, 2021. Due to a lack of government funding, only DSSR Pro is offered for academic and commercial usage as outlined above. For further information, please visit the CTV DSSR website.
Xiang-Jun
Quote from: Louis
I suggest you summarize the content of the [CTV] page in a forum post, to provide a reliable source of information about DSSR licensing, maybe?
There are two types of DSSR Pro licenses, as shown below:
- Commercial users may inquire about pricing and licensing terms by emailing techtransfer@columbia.edu (and CC to xiangjun@x3dna.org).
Academic users can obtain DSSR Pro for a one-time fee of $1000, which includes a comprehensive user manual and one year of developer support as set forth in the license agreement. Please contact techtransfer@columbia.edu (and CC to xiangjun@x3dna.org). DSSR Pro has completely superseded 3DNA, and is being continuously improved.
The free DSSR Basic academic license is no longer available as of November 11, 2021. Due to a lack of government funding, only DSSR Pro is offered for academic and commercial usage as outlined above. For further information, please visit the CTV DSSR website.
Xiang-Jun
10
General discussions (Q&As) / MOVED: Color each block nucleotide at each locations
« on: May 26, 2021, 04:16:46 pm »11
General discussions (Q&As) / MOVED: Stacking interactions in G-quadruplexes
« on: May 21, 2021, 11:48:32 am »12
Site announcements / Video: an overview of DSSR
« on: May 01, 2021, 01:32:37 pm »
I've just released a video "An overview of DSSR" -- http://docs.x3dna.org/dssr-overview/.
DSSR already has a large user base. Based on my observation, however, DSSR is still heavily underused for what it has to offer. This DSSR overview video is for new DSSR users, as well as existing ones.
As always, I appreciate your feedback.
Best regards,
Xiang-Jun
DSSR already has a large user base. Based on my observation, however, DSSR is still heavily underused for what it has to offer. This DSSR overview video is for new DSSR users, as well as existing ones.
As always, I appreciate your feedback.
Best regards,
Xiang-Jun
14
w3DNA -- web interface to 3DNA / MOVED: composite DNA template length
« on: February 18, 2021, 11:09:00 am »15
General discussions (Q&As) / MOVED: Extending DNA in protein-DNA complex - issue with final steps
« on: February 18, 2021, 11:07:23 am »16
General discussions (Q&As) / MOVED: Circular DNA parameters
« on: February 18, 2021, 11:07:07 am »17
Site announcements / DSSR-PyMOL schematics recommended in Faculty Opinions
« on: August 31, 2020, 11:31:48 am »
Recently, while visiting the NAR website on DSSR-enabled innovative schematics of 3D nucleic acid structures with PyMOL, I noticed a big red circle ① near “View Metrics”. The symbol is very obvious and a bit 'alarming'. I was curious to see what it meant. After a few clicks, I was delighted to read the following recommendation in Faculty Opinions by Quentin Vicens:
The DSSR-PyMOL schematics paper/website has been rated “Very Good”, and classified as “Good for Teaching”. See Vicens Q: Faculty Opinions Recommendation of [Lu XJ, Nucleic Acids Res 2020 48(13):e74]. In Faculty Opinions, 14 Aug 2020; 10.3410/f.738001682.793577327. A screenshot is attached below.
Quote
I really enjoyed “playing” with the revised and expanded version of Dissecting the Spatial Structure of RNA (DSSR) described by Xiang-Jun Lu in this July issue of NAR. The software is known to generate ‘block view’ representations of nucleic acids that make many parameters more immediately visible, such as base composition, stacking, and groove depth. This new version includes Watson-Crick pairs shown as single rectangles, and G quadruplexes as large squares, making such regions more quickly distinguishable from other regions within an overall tertiary structure. I was amazed at how simple and effective the web interface was, and I liked the possibility to download a PyMOL session to look at molecules under different angles. If need be, blocks can be further edited in PyMOL using the provided plugin (see on page 35). I highly recommend it!
The DSSR-PyMOL schematics paper/website has been rated “Very Good”, and classified as “Good for Teaching”. See Vicens Q: Faculty Opinions Recommendation of [Lu XJ, Nucleic Acids Res 2020 48(13):e74]. In Faculty Opinions, 14 Aug 2020; 10.3410/f.738001682.793577327. A screenshot is attached below.
18
Site announcements / DSSR 2.0 is licensed by Columbia University
« on: August 24, 2020, 08:35:04 am »
DSSR 2.0 is out. It integrates an unprecedented set of features into one computational tool, including analysis/annotation, schematic visualization, and model building of 3D nucleic acid structures. DSSR 2.0 supersedes 3DNA 2.4, which is still maintained but no additional features other than bug fixes are scheduled. See the DSSR 2.0 overview PDF.
DSSR delivers a great user experience by solving problems and saving time. Considering its usability, interoperability, features, and support, DSSR easily stands out among 'competitors'. It exemplifies a 'solid software product'. I strive to make DSSR a pragmatic tool that the structural bioinformatics community can count on.
DSSR 2.0 is licensed by Columbia University. The software remains free for academic users, with the basic user manual. The professional user manual (over 230 pages, including 7 appendices) is available for paid academic users or commercial users only. Licensing revenue helps ensure the long-term sustainability of the DSSR project.
Additionally, the paper "DSSR-enabled innovative schematics of 3D nucleic acid structures with PyMOL" has recently been published in Nucleic Acids Research, 48(13):e74. Check the web interface.
The DSSR-PyMOL paper/website has been rated "very good" and classified as "Good for Teaching". See Vicens Q: Faculty Opinions Recommendation of [Lu XJ, Nucleic Acids Res 2020 48(13):e74]. In Faculty Opinions, 14 Aug 2020; 10.3410/f.738001682.793577327.
DSSR delivers a great user experience by solving problems and saving time. Considering its usability, interoperability, features, and support, DSSR easily stands out among 'competitors'. It exemplifies a 'solid software product'. I strive to make DSSR a pragmatic tool that the structural bioinformatics community can count on.
DSSR 2.0 is licensed by Columbia University. The software remains free for academic users, with the basic user manual. The professional user manual (over 230 pages, including 7 appendices) is available for paid academic users or commercial users only. Licensing revenue helps ensure the long-term sustainability of the DSSR project.
Additionally, the paper "DSSR-enabled innovative schematics of 3D nucleic acid structures with PyMOL" has recently been published in Nucleic Acids Research, 48(13):e74. Check the web interface.
The DSSR-PyMOL paper/website has been rated "very good" and classified as "Good for Teaching". See Vicens Q: Faculty Opinions Recommendation of [Lu XJ, Nucleic Acids Res 2020 48(13):e74]. In Faculty Opinions, 14 Aug 2020; 10.3410/f.738001682.793577327.
20
General discussions (Q&As) / MOVED: Standard Values for A-, B-, C-DNA on w3DNA
« on: April 23, 2020, 09:34:21 am »
This topic has been moved to w3DNA -- web interface to 3DNA.
http://forum.x3dna.org/index.php?topic=909.0
http://forum.x3dna.org/index.php?topic=909.0
22
General discussions (Q&As) / MOVED: How to detect very distorted base pair?
« on: January 20, 2020, 11:42:18 pm »23
Site announcements / DSSR-enhanced visualization of nucleic acid structures in PyMOL
« on: December 02, 2019, 12:33:46 pm »
This website http://skmatic.x3dna.org/ (see screenshot below) aims to showcase DSSR-enabled cartoon-block schematics of nucleic acid structures using PyMOL. It presents a simple interface to browse pre-calculated PDB entries with a set of default settings: long rectangular blocks for Watson-Crick base-pairs, square blocks for G-tetrads in G-quadruplexes, with minor-groove edges in black. Users can also specify an URL to a PDB- or mmCIF-formatted file or upload such an atomic coordinates file directly, and set several common options to customerize to the rendered image.
Moreover, a web API to DSSR-PyMOL schematics is available to allow for its easy integration into third-party tools.

Input a PDB id
Pre-calculated cartoon-block images together with summary information are available for PDB entries of nucleic-acid-containing structures. Note that gigantic structures like ribosomes that are only represented in mmCIF format are excluded from the resource. The base block images are most effective for small to medium-sized structures.
Here are a few examples:
Each entry is shown with images in six orthogonal perspectives: front, back, right, left, top, bottom. The 'front' image (upper-left in the panel) is oriented into the most-extended view with the DSSR --blocview option. The corresponding PyMOL session file and PDB coordinate file are available for download. One can also visualize the structure interactively via 3Dmol.js.
Sample PDB entries
Users can browse random samples of pre-calculated PDB entries. The number should be between 3 and 99, with a default of 12 entries (see below for an example). Simply click the 'Submit' button or the link "Random samples (3 to 99)" to see random results of 12 entries each time.
Specify an coordinate file
The atomic coordinate file must be in PDB or mmCIF format, and can be optionally gzipped (.gz). One can either specify an URL to or select a coordinate file. Several common options are available to allow for user customizations.
Web API help message
Sample images
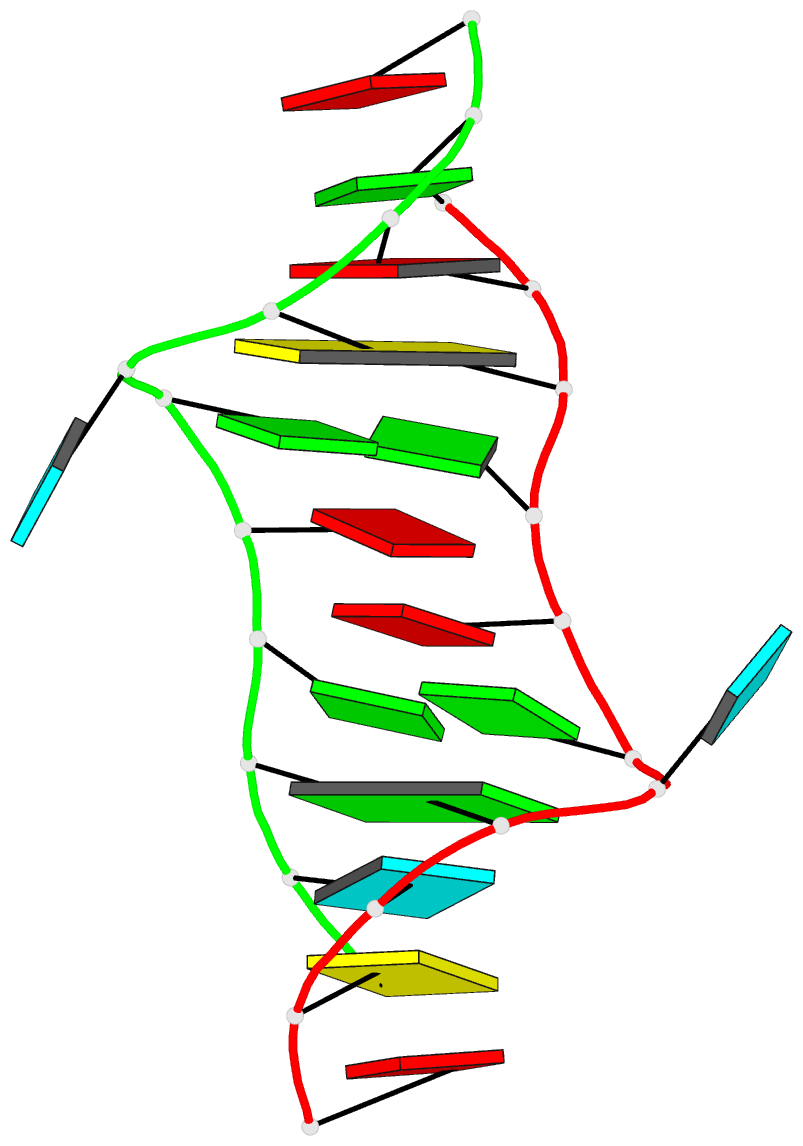
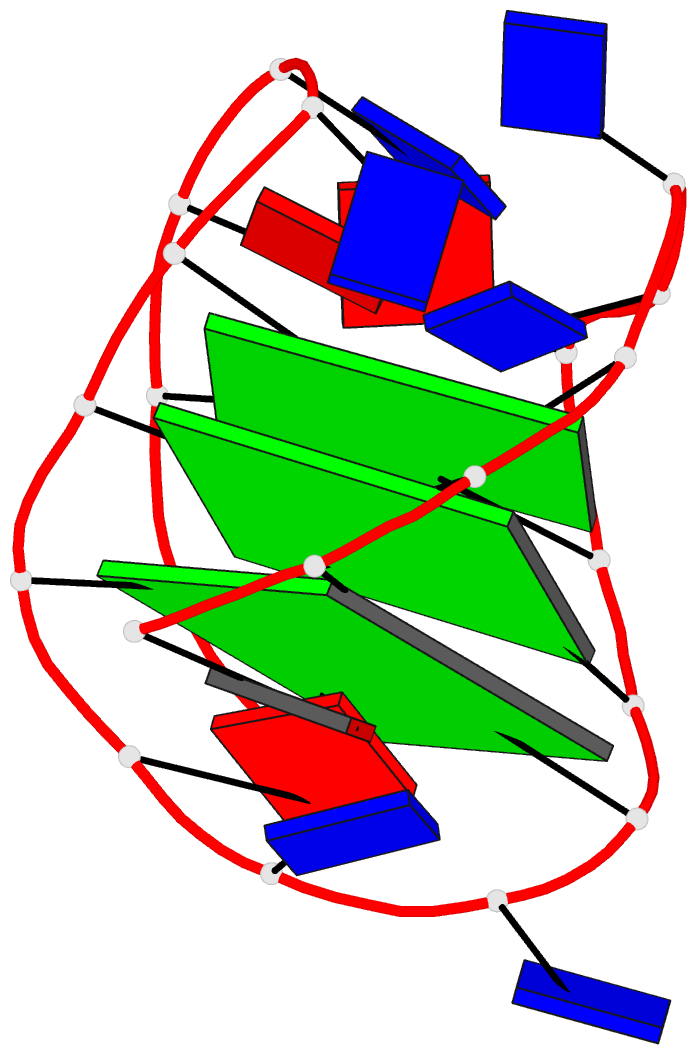
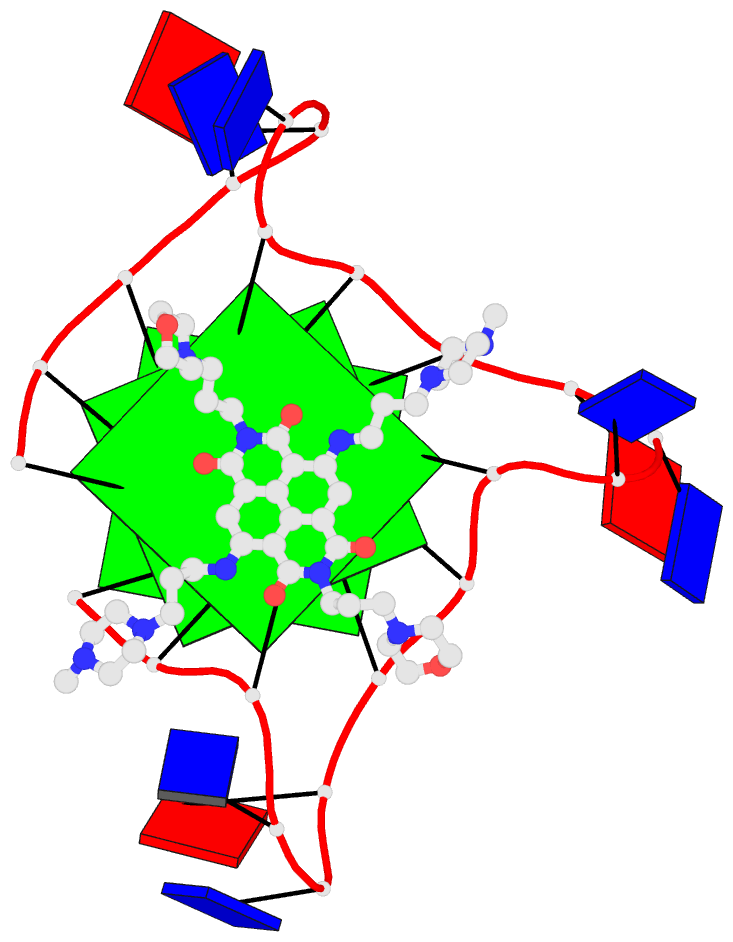
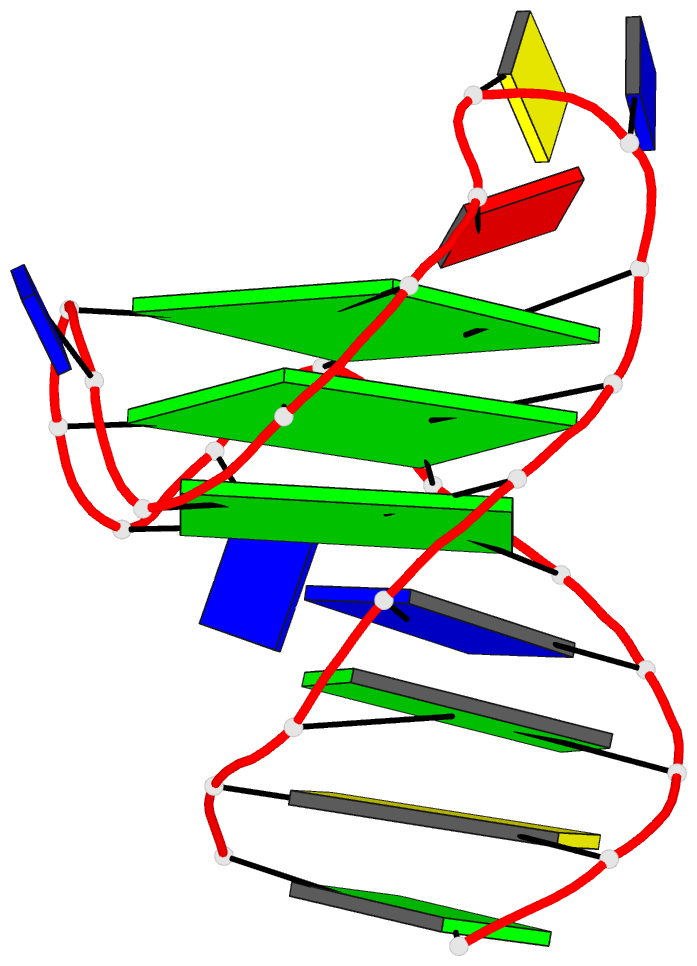
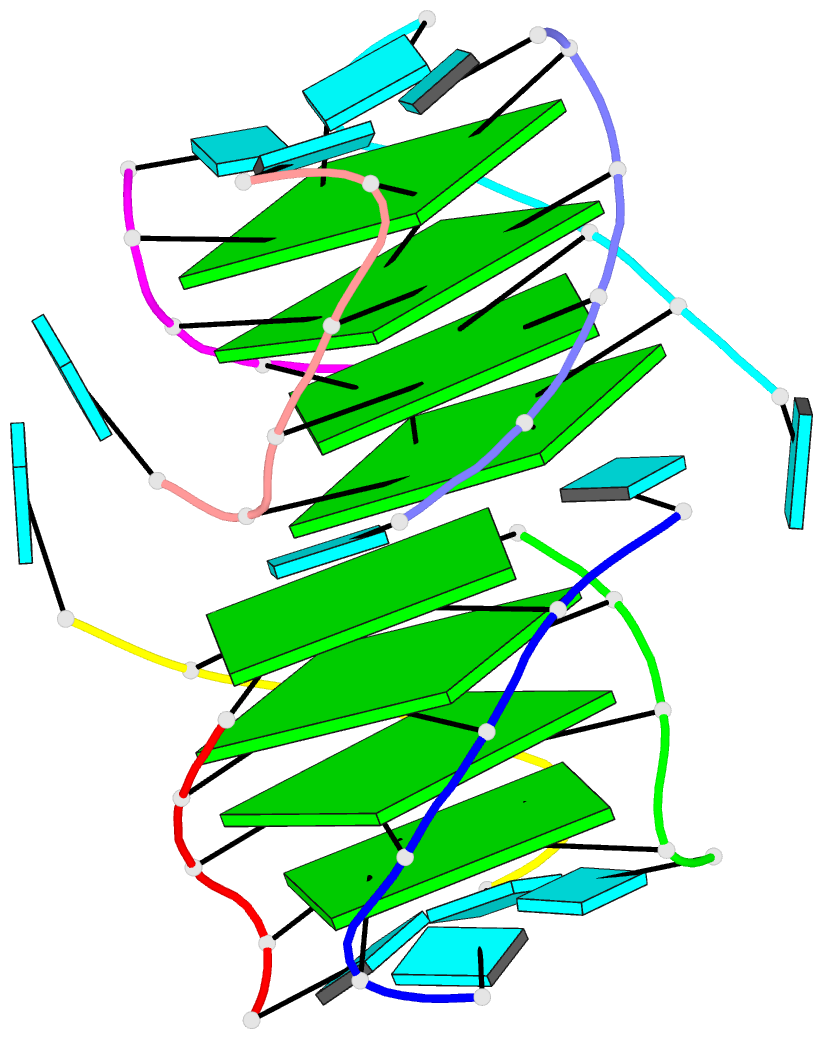
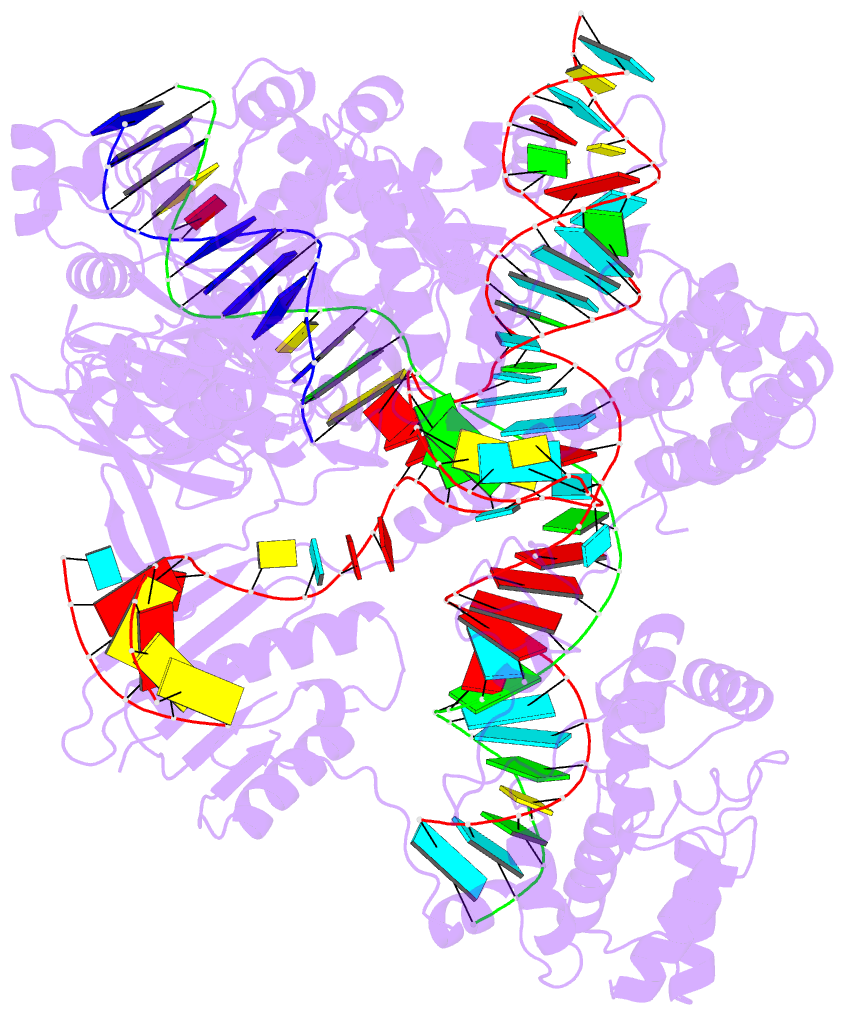
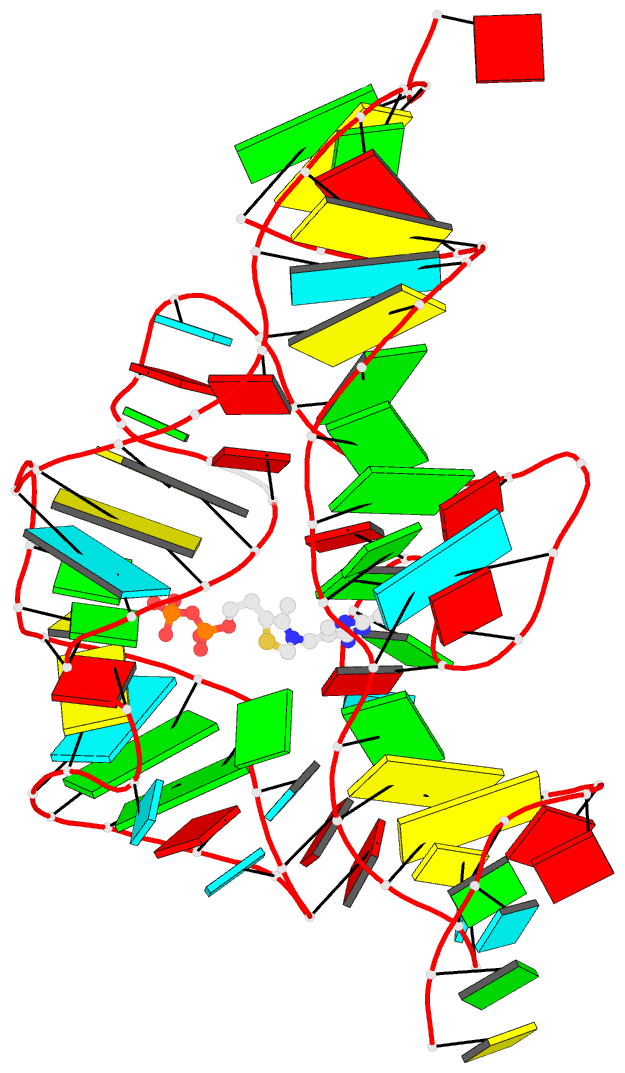
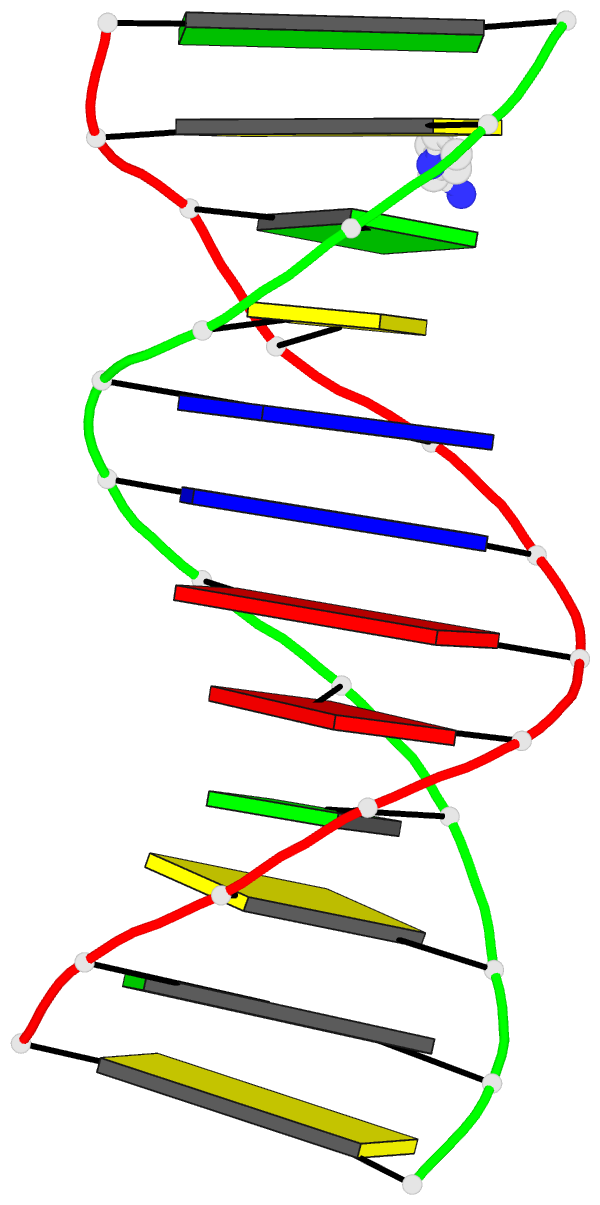
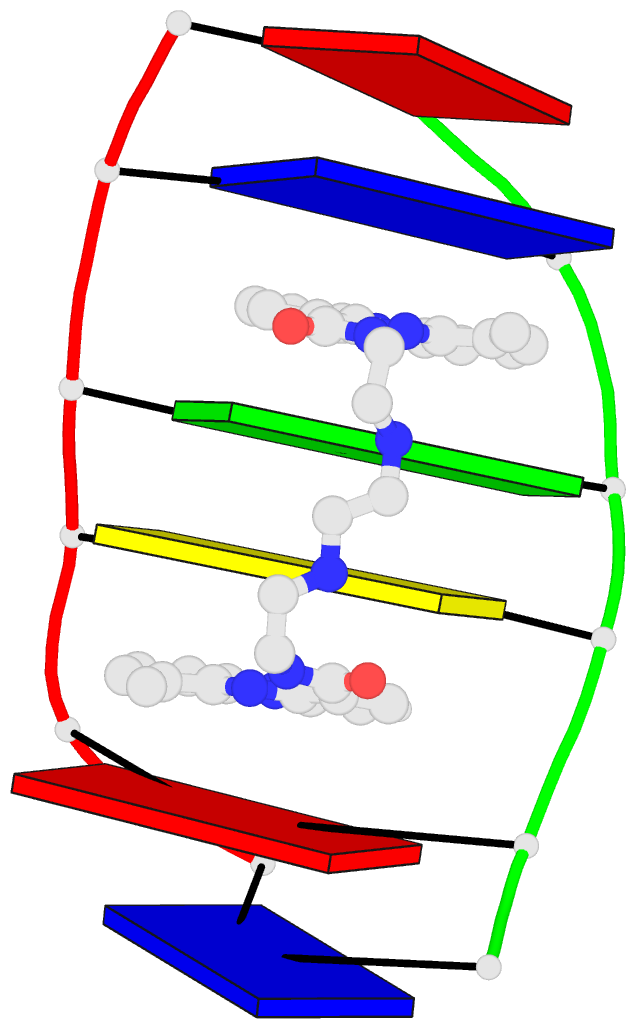
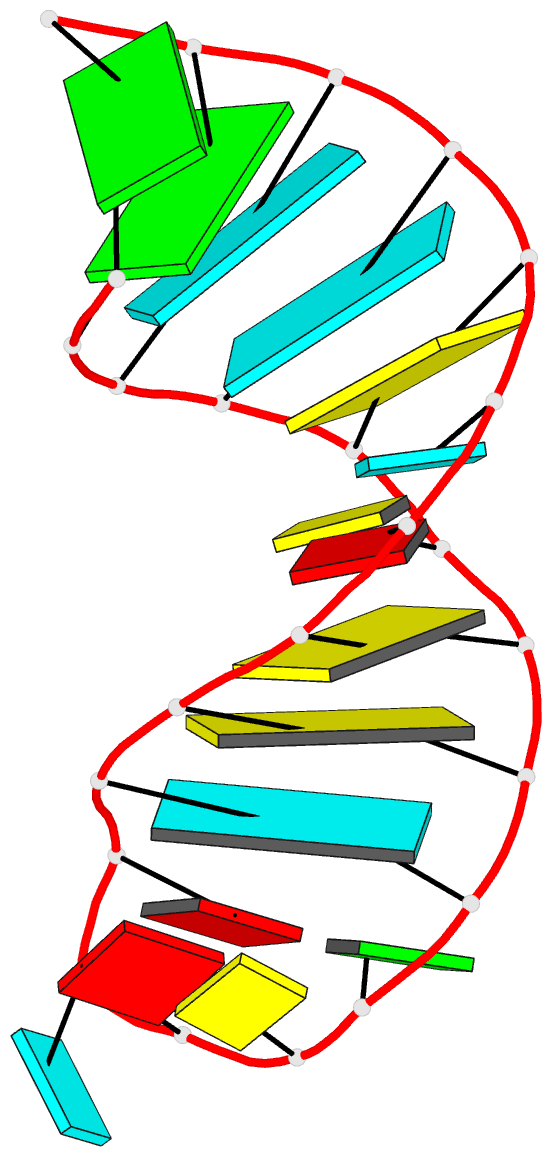
Moreover, a web API to DSSR-PyMOL schematics is available to allow for its easy integration into third-party tools.
Input a PDB id
Pre-calculated cartoon-block images together with summary information are available for PDB entries of nucleic-acid-containing structures. Note that gigantic structures like ribosomes that are only represented in mmCIF format are excluded from the resource. The base block images are most effective for small to medium-sized structures.
Here are a few examples:
- 1ehz, the crystal structure of yeast phenylalanine trna at 1.93-A resolution
- 2lx1, the major conformation of the internal loop 5'GAGU/3'UGAG
- 2grb, the crystal structure of an RNA quadruplex containing inosine-tetrad
- 4da3, the crystal structure of an intramolecular human telomeric DNA G-quadruplex 21-mer bound by the naphthalene diimide compound MM41
- 1oct, crystal structure of the Oct-1 POU domain bound to an octamer site
- 2hoj, the crystal structure of an E. coli thi-box riboswitch bound to thiamine pyrophosphate, manganese ions
Each entry is shown with images in six orthogonal perspectives: front, back, right, left, top, bottom. The 'front' image (upper-left in the panel) is oriented into the most-extended view with the DSSR --blocview option. The corresponding PyMOL session file and PDB coordinate file are available for download. One can also visualize the structure interactively via 3Dmol.js.
Sample PDB entries
Users can browse random samples of pre-calculated PDB entries. The number should be between 3 and 99, with a default of 12 entries (see below for an example). Simply click the 'Submit' button or the link "Random samples (3 to 99)" to see random results of 12 entries each time.
Specify an coordinate file
The atomic coordinate file must be in PDB or mmCIF format, and can be optionally gzipped (.gz). One can either specify an URL to or select a coordinate file. Several common options are available to allow for user customizations.
Web API help message
Usage with 'http' (HTTPie):
http -f http://skmatic.x3dna.org/api [options] url=|model@
http http://skmatic.x3dna.org/api/pdb/pdb_id -- for a pre-calculated PDB entry
http http://skmatic.x3dna.org/api/help -- display this help message
Options:
block_file=styles-in-free-text-format [e.g., block_file=wc-minor]
block_color=nt-selection-and-color [e.g., block_color='A:pink']
block_depth=thickness-of-base-block [e.g., block_depth=1.2]
r3d_file=true-or-FALSE(default) [e.g., r3d_file=true]
raw_xyz=true-or-FALSE(default) [e.g., raw_xyz=true]
Required parameter
url=URL-to-coordinate-file [e.g., url=https://files.rcsb.org/download/1ehz.pdb.gz]
model@coordinate-file [e.g., model@1ehz.cif]
# Only one must be specified. 'url' precedes 'model' when both are specified.
# The coordinate file must be in PDB or PDBx/mmCIF format, optionally gzipped.
Examples
http -f http://skmatic.x3dna.org/api block_file='wc-minor' model@1ehz.cif r3d_file=t
http -f http://skmatic.x3dna.org/api url=https://files.rcsb.org/download/1ehz.pdb.gz -d -o 1ehz.png
http http://skmatic.x3dna.org/api/pdb/1ehz -d -o 1ehz.png
# with 'curl'
curl http://skmatic.x3dna.org/api -F 'model=@1msy.pdb' -F 'block_file=wc-minor' -F 'r3d_file=1'
curl http://skmatic.x3dna.org/api -F 'url=https://files.rcsb.org/download/1ehz.pdb.gz' -o 1ehz.png
curl http://skmatic.x3dna.org/api/pdb/1ehz -o 1ehz.png
Sample images
24
Feature requests / MOVED: building circular DNA
« on: July 16, 2019, 11:56:46 am »
This topic has been moved to General discussions (Q&As).
http://forum.x3dna.org/index.php?topic=862.0
http://forum.x3dna.org/index.php?topic=862.0
25
Site announcements / Web 3DNA 2.0 is highlighted in NAR web server issue 2019
« on: July 01, 2019, 07:47:39 pm »
It is a great pleasure to see that our article "Web 3DNA 2.0 for the analysis, visualization, and modeling of 3D nucleic acid structures" has been highlighted in the cover page of the web server issue of NAR’19. According to the editor, This year, 331 proposals were submitted and 122, or 37%, were approved for manuscript submission. Of those approved, 94, or 77%, were ultimately accepted for publication. Overall, that corresponds to a ~28% acceptance rate.
The cover image and its caption are shown below. Moreover, details on how the cover image was created are available on the 3DNA Forum.
 Caption: Examples of customized molecular models that can be generated with 3DNA: (top) a chromatin-like, nucleosome-decorated DNA with the structures of known histone-DNA assemblies placed at user-defined binding sites; (lower left) molecular schematic of a DNA trinucleotide diphosphate illustrating the base planes and reference frames used to construct and analyze 3D nucleic acid-containing structures; (lower right) customized single-stranded tRNA model built from a user-defined base sequence and a set of rigid-body parameters describing the desired placement of successive bases. Color code of base blocks: A, red; C, yellow; G, green; T, blue; U, cyan.
Caption: Examples of customized molecular models that can be generated with 3DNA: (top) a chromatin-like, nucleosome-decorated DNA with the structures of known histone-DNA assemblies placed at user-defined binding sites; (lower left) molecular schematic of a DNA trinucleotide diphosphate illustrating the base planes and reference frames used to construct and analyze 3D nucleic acid-containing structures; (lower right) customized single-stranded tRNA model built from a user-defined base sequence and a set of rigid-body parameters describing the desired placement of successive bases. Color code of base blocks: A, red; C, yellow; G, green; T, blue; U, cyan.
The cover image and its caption are shown below. Moreover, details on how the cover image was created are available on the 3DNA Forum.
Funded by the NIH R24GM153869 grant on X3DNA-DSSR, an NIGMS National Resource for Structural Bioinformatics of Nucleic Acids
Created and maintained by Dr. Xiang-Jun Lu, Department of Biological Sciences, Columbia University