Okay, let's check step-by-step how
find_pair is working for your two attached structures.
- The output for 30ns@mod_2.pdb is as you expected.
find_pair 30ns@mod_2.pdb 30ns@mod_2.bps
more 30ns@mod_2.bps
# the output is as below
30ns@mod_2.pdb
30ns@mod_2.out
2 # duplex
14 # number of base-pairs
1 1 # explicit bp numbering/hetero atoms
1 31 0 # 1 | ....>-:...1_:[DA5]a-**--t[TPN]:..31_:-<.... 4.71 1.82 45.14 7.30 7.61
2 30 0 # 2 | ....>-:...2_:[.DA]A-----t[TPN]:..30_:-<.... 0.51 0.30 13.22 9.70 -3.24
3 29 0 # 3 | ....>-:...3_:[.DT]T-----a[APN]:..29_:-<.... 0.73 0.73 8.66 9.13 -2.38
4 28 0 # 4 | ....>-:...4_:[.DT]T-----a[APN]:..28_:-<.... 0.19 0.09 13.10 9.21 -3.98
5 27 0 # 5 | ....>-:...5_:[.DT]T-----a[APN]:..27_:-<.... 1.03 1.03 17.68 8.93 0.96
6 26 0 # 6 | ....>-:...6_:[.DT]T-----a[APN]:..26_:-<.... 0.23 0.22 16.23 9.13 -3.52
7 25 0 # 7 | ....>-:...7_:[.DT]T-----a[APN]:..25_:-<.... 0.37 0.36 10.91 8.99 -3.36
8 24 0 # 8 | ....>-:...8_:[.DT]T-----a[APN]:..24_:-<.... 0.30 0.01 9.28 9.21 -4.21
9 23 0 # 9 | ....>-:...9_:[.DT]T-----a[APN]:..23_:-<.... 0.49 0.12 10.87 9.24 -3.73
10 22 0 # 10 | ....>-:..10_:[.DT]T-----a[APN]:..22_:-<.... 0.18 0.05 14.36 8.97 -4.00
11 21 0 # 11 | ....>-:..11_:[.DA]A-----t[TPN]:..21_:-<.... 0.47 0.21 1.26 9.28 -4.05
12 20 0 # 12 | ....>-:..12_:[.DT]T-----a[APN]:..20_:-<.... 0.34 0.30 5.05 9.10 -3.80
13 19 0 # 13 | ....>-:..13_:[.DT]T-----a[APN]:..19_:-<.... 0.46 0.45 22.22 8.98 -2.53
14 18 0 # 14 | ....>-:..14_:[.DT]T-----a[APN]:..18_:-<.... 0.47 0.28 25.44 9.34 -2.71
##### Base-pair criteria used: 4.00 0.00 15.00 2.50 65.00 4.50 7.50 [ O N]
##### 1 non-Watson-Crick base-pair, and 1 helix (0 isolated bps)
##### Helix #1 (14): 1 - 14 ***broken O3' to P[i+1] linkage***
Using Jmol/PyMOL/RasMol to visualize the structure, one can easily verify that find_pair is behaving properly. - Now repeat the procedute for 30ns_nsp_mod.pdb, you'd get the following result which you thought as "improper":
find_pair 30ns_nsp_mod.pdb 30ns_nsp_mod.bps
more 30ns_nsp_mod.bps
# the output is as below
30ns_nsp_mod.pdb
30ns_nsp_mod.out
2 # duplex
10 # number of base-pairs
1 1 # explicit bp numbering/hetero atoms
1 31 0 # 1 | ....>-:...1_:[..A]A-**--T[..T]:..31_:-<.... 6.27 0.03 22.42 9.06 6.46
2 30 0 # 2 | ....>-:...2_:[..A]A-**--T[..T]:..30_:-<.... 4.14 0.42 23.12 9.56 5.13
3 29 0 # 3 | ....>-:...3_:[..T]T-----A[ADE]:..29_:-<.... 0.13 0.01 20.45 9.45 -3.83
4 28 0 # 4 | ....>-:...4_:[..T]T-----A[..A]:..28_:-<.... 0.28 0.28 19.08 9.01 -3.21
5 27 0 # 5 | ....>-:...5_:[..T]T-----A[..A]:..27_:-<.... 0.40 0.11 18.15 9.35 -3.47
6 26 0 # 6 | ....>-:...6_:[..T]T-----A[ADE]:..26_:-<.... 0.64 0.51 27.99 9.05 -1.94
7 25 9 # 7 x ....>-:...7_:[..T]T-----A[..A]:..25_:-<.... 0.62 0.56 25.16 9.15 -2.00
10 22 1 # 8 + ....>-:..10_:[..T]T-----A[..A]:..22_:-<.... 0.64 0.53 15.83 9.01 -2.50
13 19 0 # 9 | ....>-:..13_:[..T]T-----A[ADE]:..19_:-<.... 0.48 0.19 18.51 5.17 -3.21
14 18 0 # 10 | ....>-:..14_:[..T]T-----A[..A]:..18_:-<.... 0.27 0.26 30.07 4.91 -2.71
##### Base-pair criteria used: 4.00 0.00 15.00 2.50 65.00 4.50 7.50 [ O N]
##### 2 non-Watson-Crick base-pairs, and 3 helices (1 isolated bp)
##### Helix #1 (7): 1 - 7 ***broken O3' to P[i+1] linkage***
##### Helix #2 (1): 8
##### Helix #3 (2): 9 - 10 ***broken O3' to P[i+1] linkage***
As shown in the image below, find_pair is again behaving as it should for this case. Is this structure itself what you'd expect?
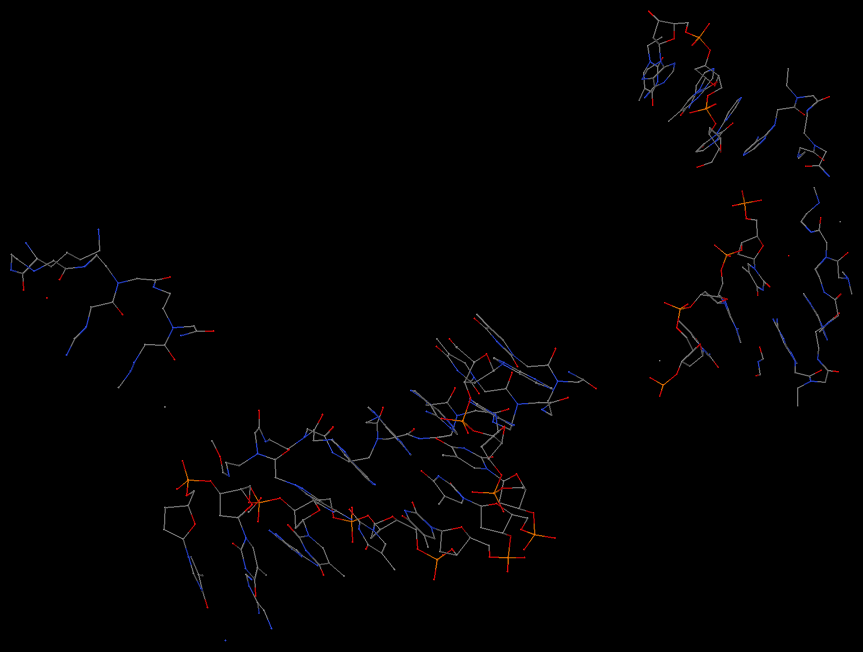
In my experience, whenever a user suspects the output from
find_pair as "improper", it is more than likely that the structure itself is "weird" -- if in doubt,
always check your structure using a molecular graphics visualization program (Jmol/PyMOL/RasMol etc). Of course, I am consistently on the watch to refine
find_pair, especially for the edge cases.
Xiang-Jun


 Topic: Analysis of PDB file:query (Read 135687 times)
Topic: Analysis of PDB file:query (Read 135687 times)