I am excited to announce that a paper titled "
DSSR-enhanced visualization of nucleic acid structures in Jmol" has just been published online for the 2017 web-server issue of
Nucleic Acids Research (NAR). Co-authored by Robert Hanson and me, this paper represents an idealized result I could expect from a scientific collaboration. I first approached Bob in October 2013 at a meeting organized by RCSB PDB at Rutgers, and we then met again in July 2014 in Paris. Over the years, we have communicated extensively via email, facilitated by Skype. Collaborating with Bob has been a truly exciting experience, and it is gratifying to see a joint publication coming out of our efforts.
The DSSR-Jmol integration bridges the DSSR command-line analyzing tool and the Jmol molecular viewer seamlessly together via a simple JSON interface and a powerful query language. This work fills a gap in RNA structural bioinformatics, and brings the 3D interactive visualization of nucleic acid structures to an entirely new level. The website (
http://jmol.x3dna.org) is fully functional, useful to researchers, educators, and students alike. Furthermore, it can serve as a starting point for anyone who wishes to develop additional interactive web-based resources involving nucleic acid structures.
The DSSR-Jmol paper has been featured in the cover image of the NAR'17 web-server issue. Undoubtedly, this recognition would further increase this publicity of this solid piece of work. (note added on June 30, 2017)
The abstract of the paper is quoted below:
Sophisticated and interactive visualizations are essential for making sense of the intricate 3D structures of macromolecules. For proteins, secondary structural components are routinely featured in molecular graphics visualizations. However, the field of RNA structural bioinformatics is still lagging behind; for example, current molecular graphics tools lack built-in support even for base pairs, double helices, or hairpin loops. DSSR (Dissecting the Spatial Structure of RNA) is an integrated and automated command-line tool for the analysis and annotation of RNA tertiary structures. It calculates a comprehensive and unique set of features for characterizing RNA, as well as DNA structures. Jmol is a widely used, open-source Java viewer for 3D structures, with a powerful scripting language. JSmol, its reincarnation based on native JavaScript, has a predominant position in the post Java-applet era for web-based visualization of molecular structures. The DSSR-Jmol integration presented here makes salient features of DSSR readily accessible, either via the Java-based Jmol application itself, or its HTML5-based equivalent, JSmol. The DSSR web service accepts 3D coordinate files (in mmCIF or PDB format) initiated from a Jmol or JSmol session and returns DSSR-derived structural features in JSON format. This seamless combination of DSSR and Jmol/JSmol brings the molecular graphics of 3D RNA structures to a similar level as that for proteins, and enables a much deeper analysis of structural characteristics. It fills a gap in RNA structural bioinformatics, and is freely accessible (via the Jmol application or the JSmol-based website http://jmol.x3dna.org).
This section on the 3DNA Forum is dedicated to topics on reproducing the results reported in the DSSR-Jmol article,
and the cover image. Scripts and related data files where necessary are provided so interested parties can rigorously reproduce our results. We welcome
any questions and comments you may have. Please post them here instead of (or in addition to) sending me emails.
Note that the reported results in the paper were based on
DSSR version 1.6.8 (released on 2017-03-28) and
Jmol version 14.13.1 (released on 2017-04-09). During the proof stage, Jmol was updated to version
14.15.1 (released on 2017-04-27), which was the one reported in the manuscript and the supplementary data.
Best regards,
Xiang-Jun
For completeness, here are Figure 1 (brief description of DSSR algorithms) and Figure 2 (screenshot of the DSSR-Jmol website).

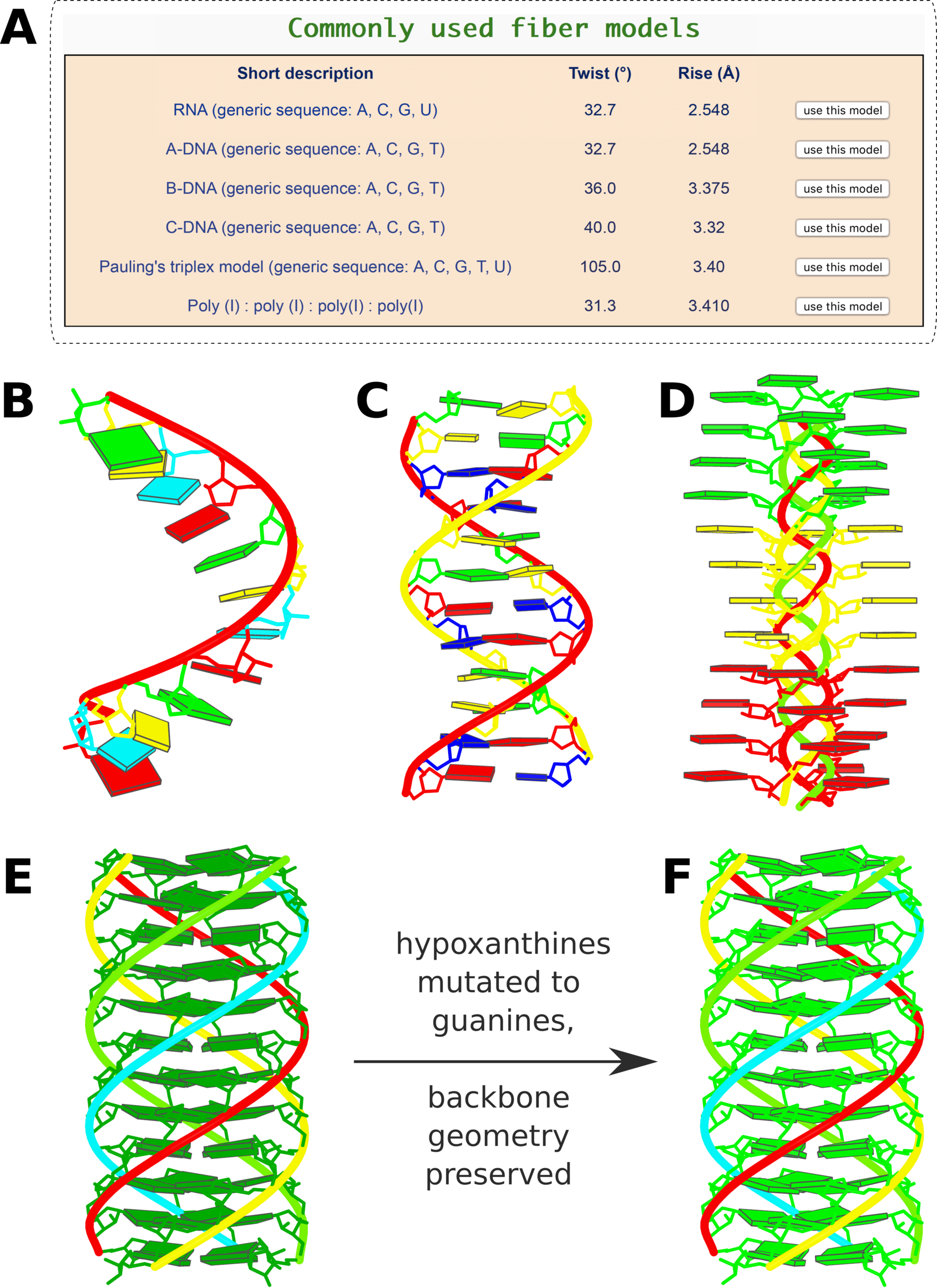
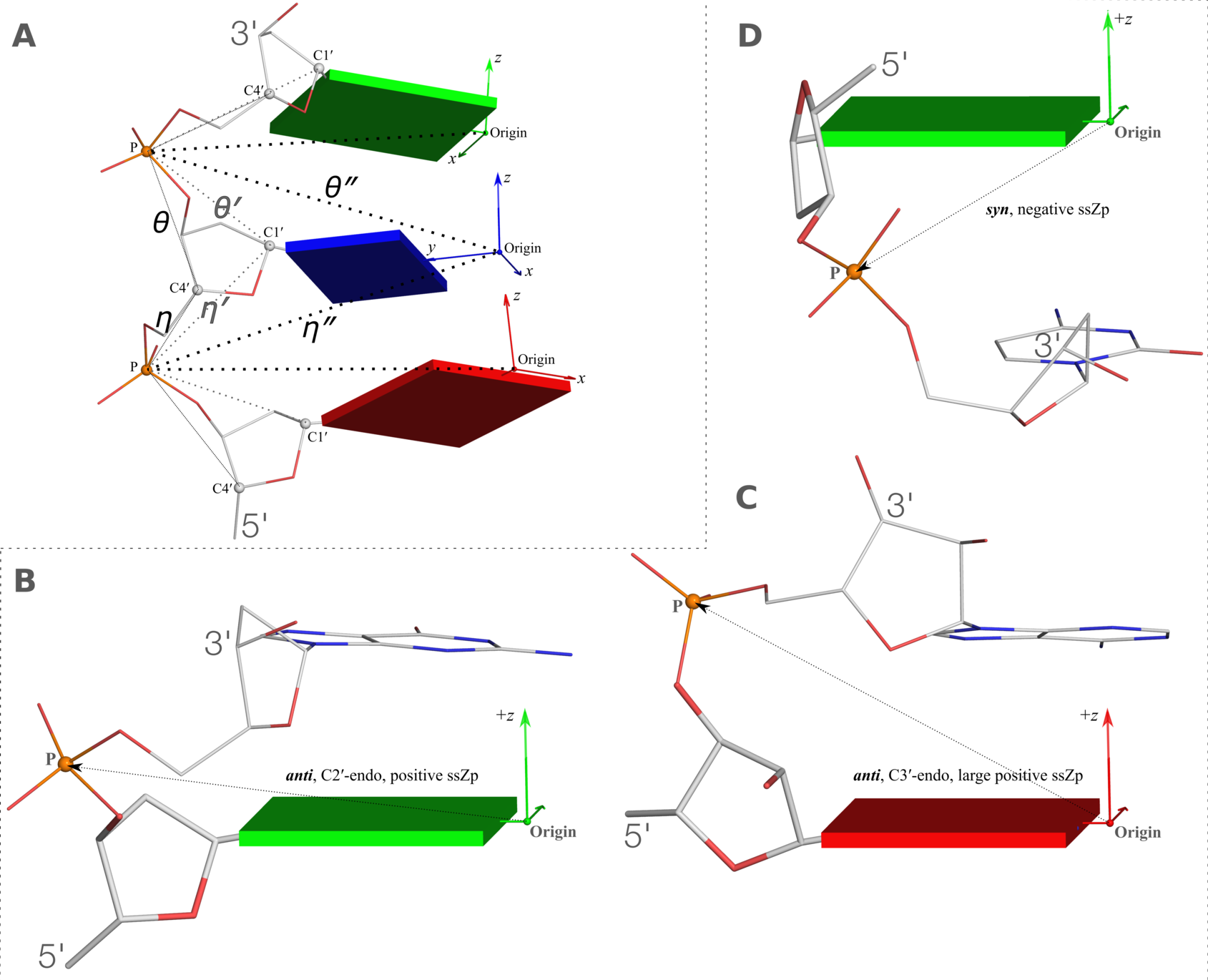
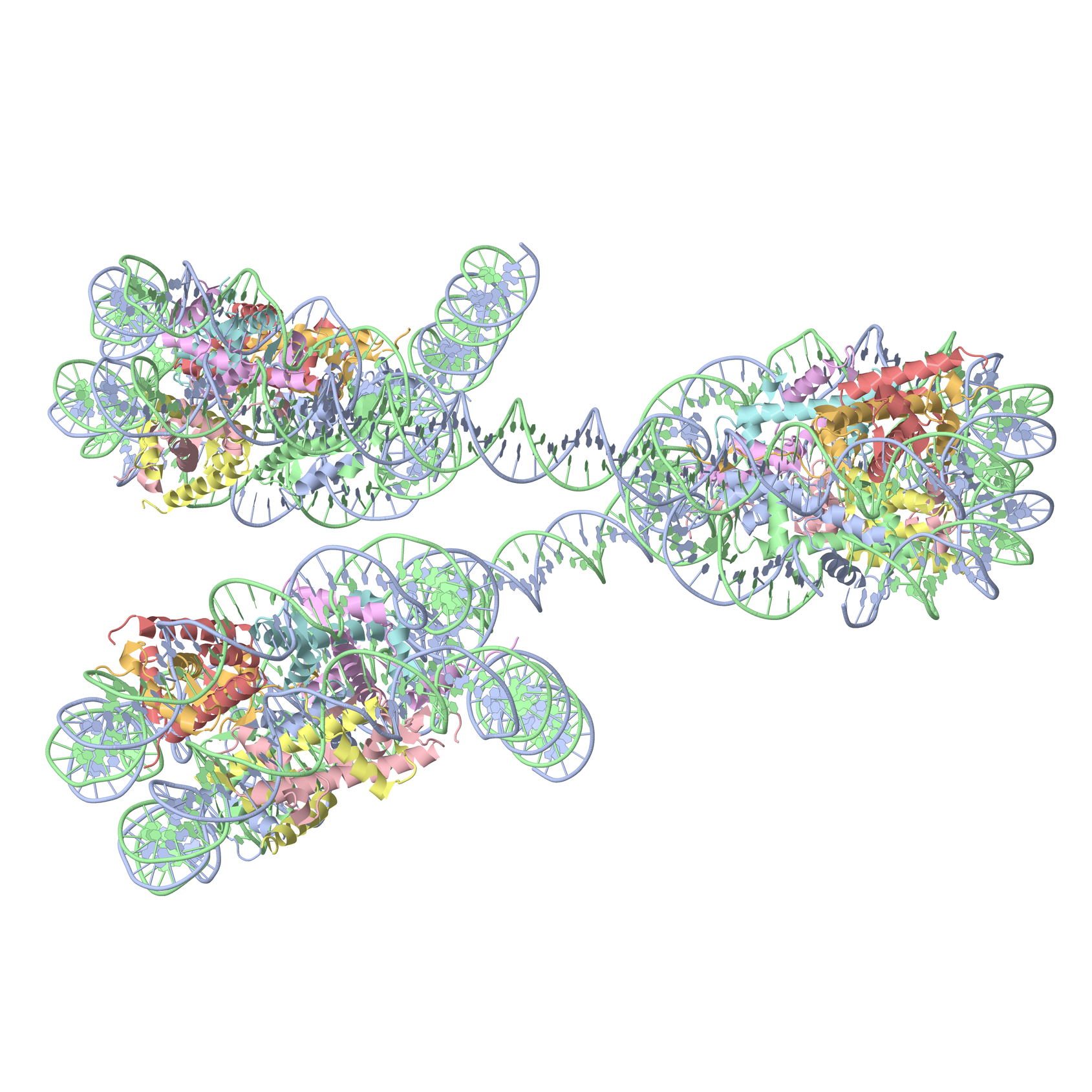
Definitions of key nucleic acid structural components in DSSR [reproduced from Figure 1 of reference (9)]. (A) Nucleotides are recognized using standard atom names and base planarity. This method works for both the standard bases (A, C, G, T and U), and those of modified nucleotides, regardless of their tautomeric or protonation states. (B) Bases are assigned a standard reference frame (25) that is independent of sequence identity: purines and pyrimidines are symmetrically placed with respect to the sugar. (C) The standard base frame is derived from an idealized Watson-Crick base pair, and defines three base edges (Watson-Crick, minor groove, and Major groove) that are used to classify pairing interactions. (D) Base pairs are identified from the co-planarity of base rings and the occurrence of hydrogen bonds. This geometric algorithm can find canonical (Watson-Crick and G–U wobble) as well as non-canonical pairs. Higher-order (three or more) co-planar base associations, termed multiplets, are also detected. (E) Helices are defined by stacking interactions of base pairs, regardless of pairing type (canonical or otherwise) or backbone connectivity (covalently connected or broken). A helix consists of at least two base pairs. The same algorithm is applied to identify continuous base stacks that are outside of helical regions, by using bases instead of pairs as the assembly unit. Nucleotides not involved in base-stacking interactions are collected into one separate group. A stem is defined as a special type of helix, made up of canonical pairs and with a continuous backbone along each strand. Coaxial stacking is defined by the presence of two or more stems within one helix. An isolated canonical pair is one that is not contained within a stem. (F) ‘Closed’ loops of various types (hairpin, bulge, internal, and junction loops) are delineated by stems or isolated pairs, and specified by the lengths of the intervening, consecutive nucleotide segments. A kissing-loop motif entails formation of one or more canonical pairs between the bases in different hairpin loops. Single-stranded segments that lie outside loops are separately listed.

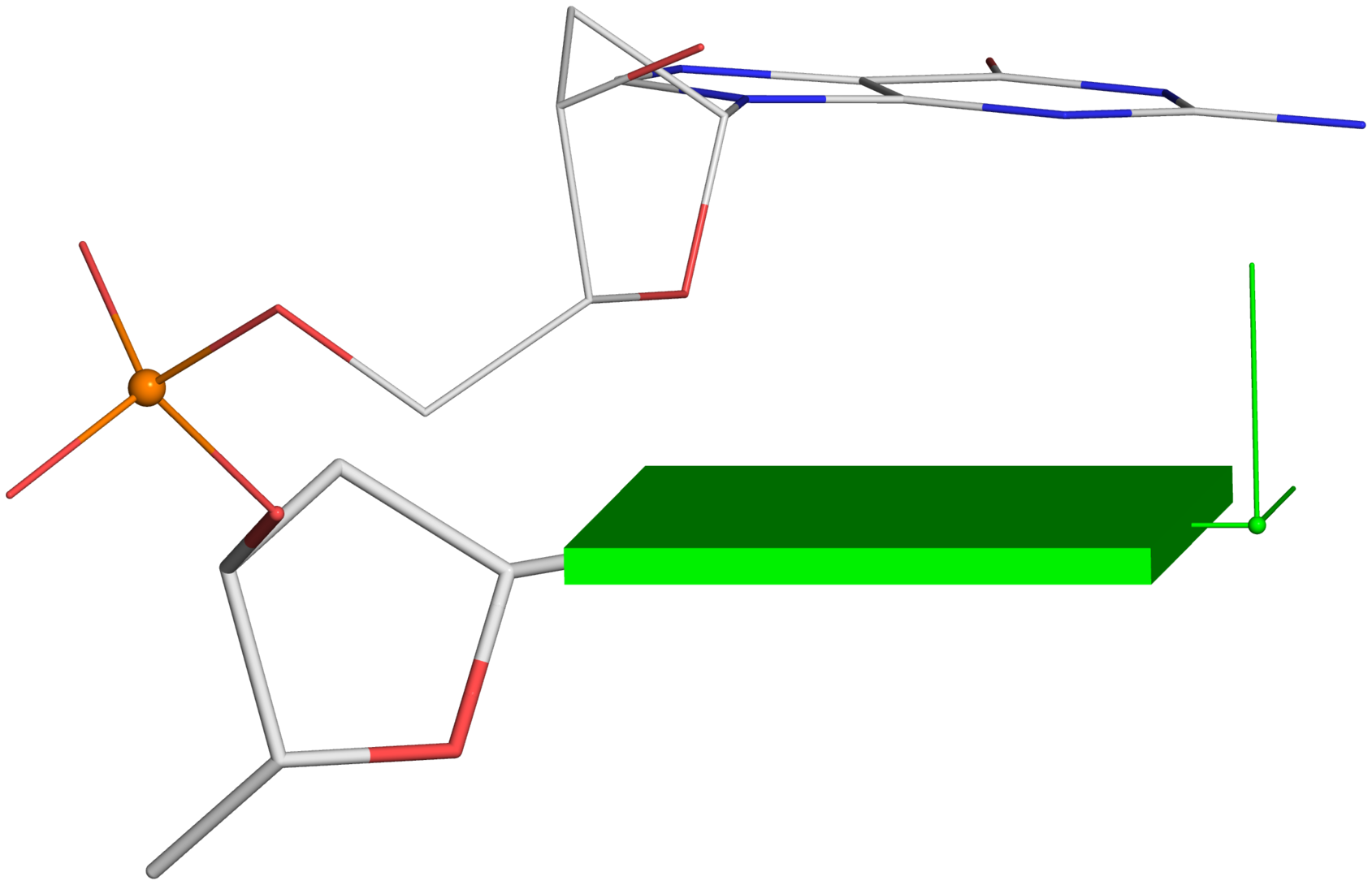
A screenshot of the DSSR-JSmol web interface, highlighting the two reverse Hoogsteen pairs (U8–A14 and 5MU54–1MA58) of yeast phenylalanine tRNA (PDB id: 1ehz). (A) DSSR-derived structural features integrated into Jmol. (B) The main JSmol viewer canvas for visualization and interactive manipulations. (C) Common representation styles for selected structural features. (D) A simple text input field for advanced users to enter (short) Jmol script commands. (E) Structure input by PDB id, file upload (drag-and-drop), or selecting one from the twelve sample RNA structures. (F) Utilities to toggle between two states for six common cases. (G) Export of coordinate file or PNG image. (H) Links to online resources for DSSR and Jmol.


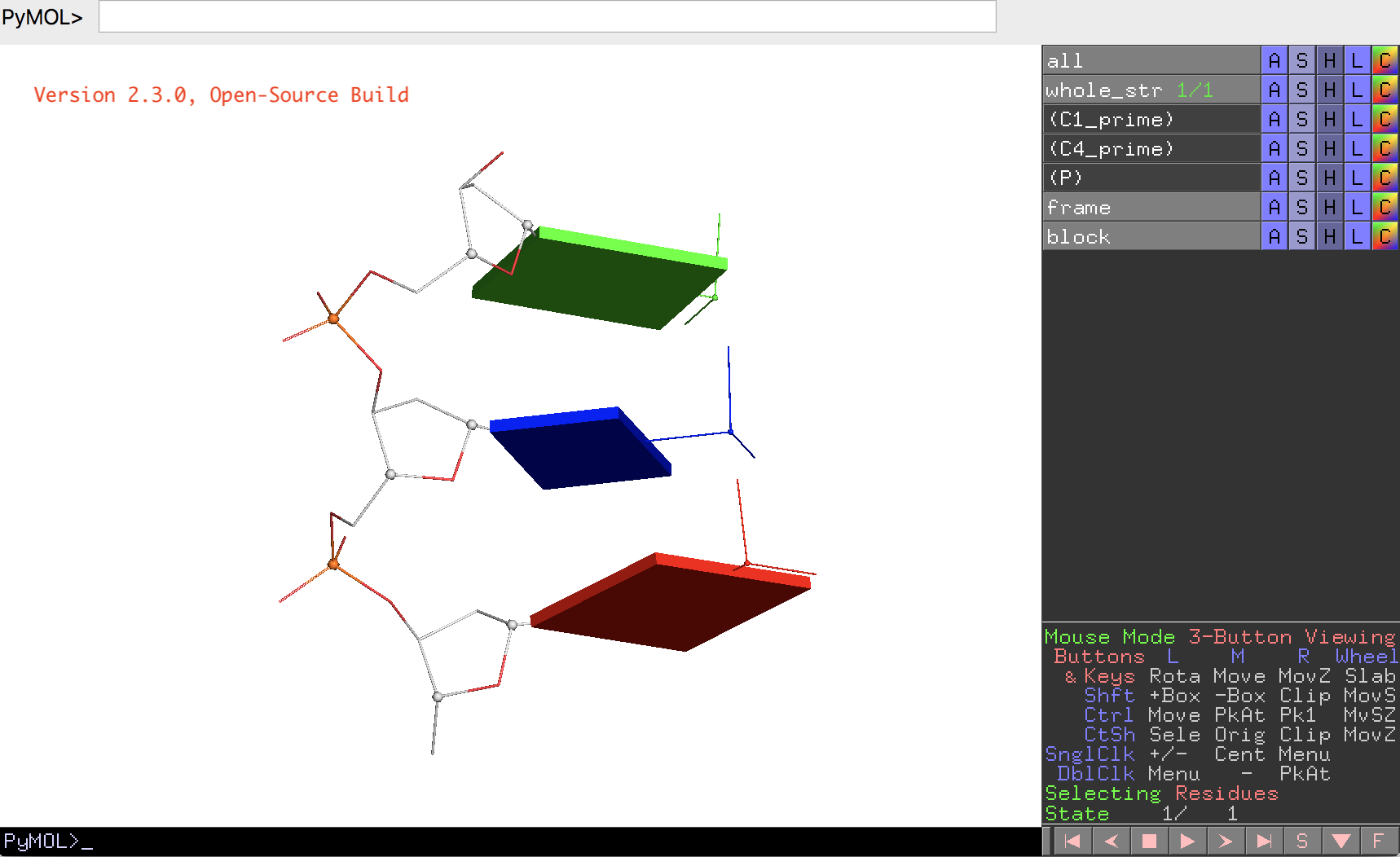
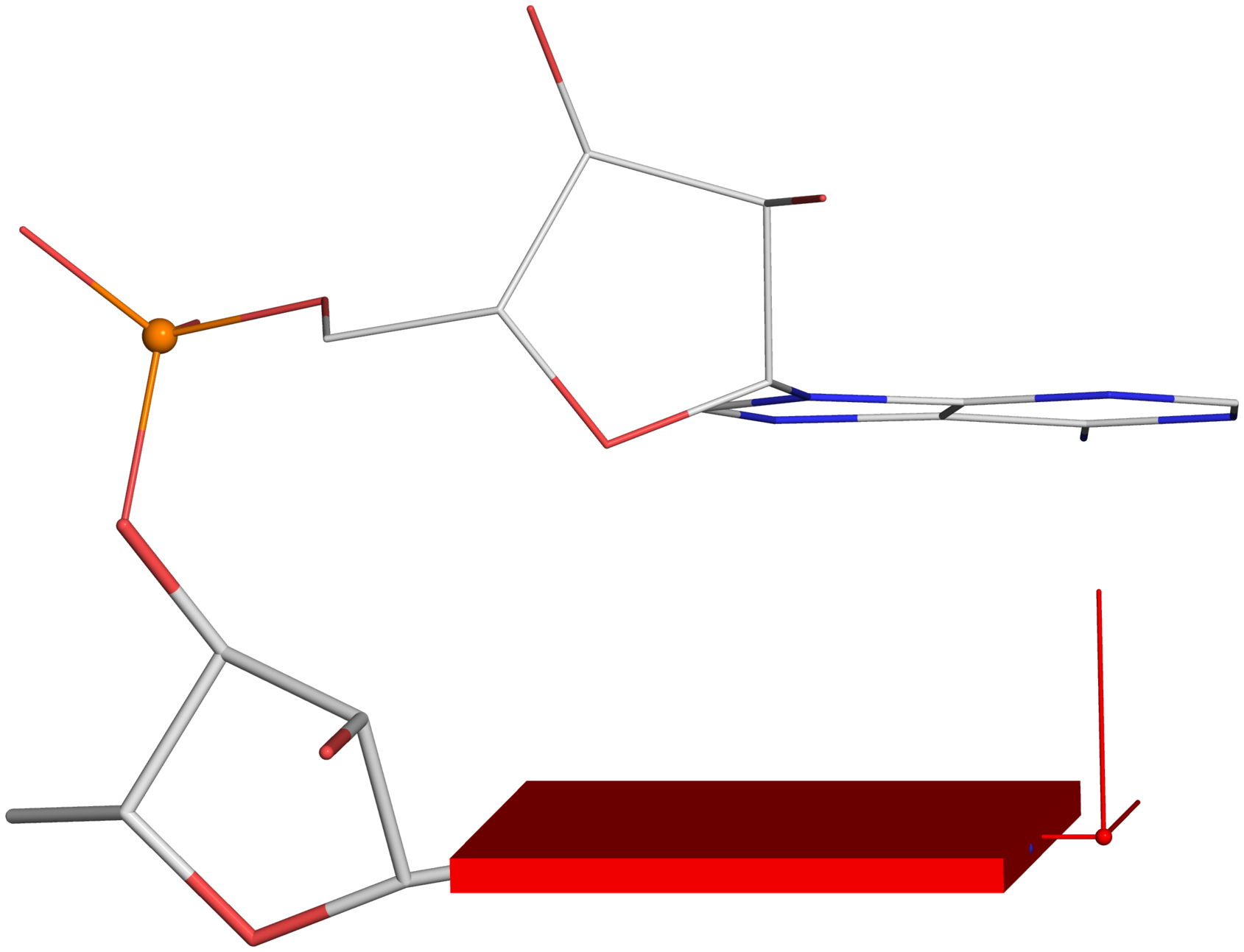
 -- Fig. 2B
-- Fig. 2B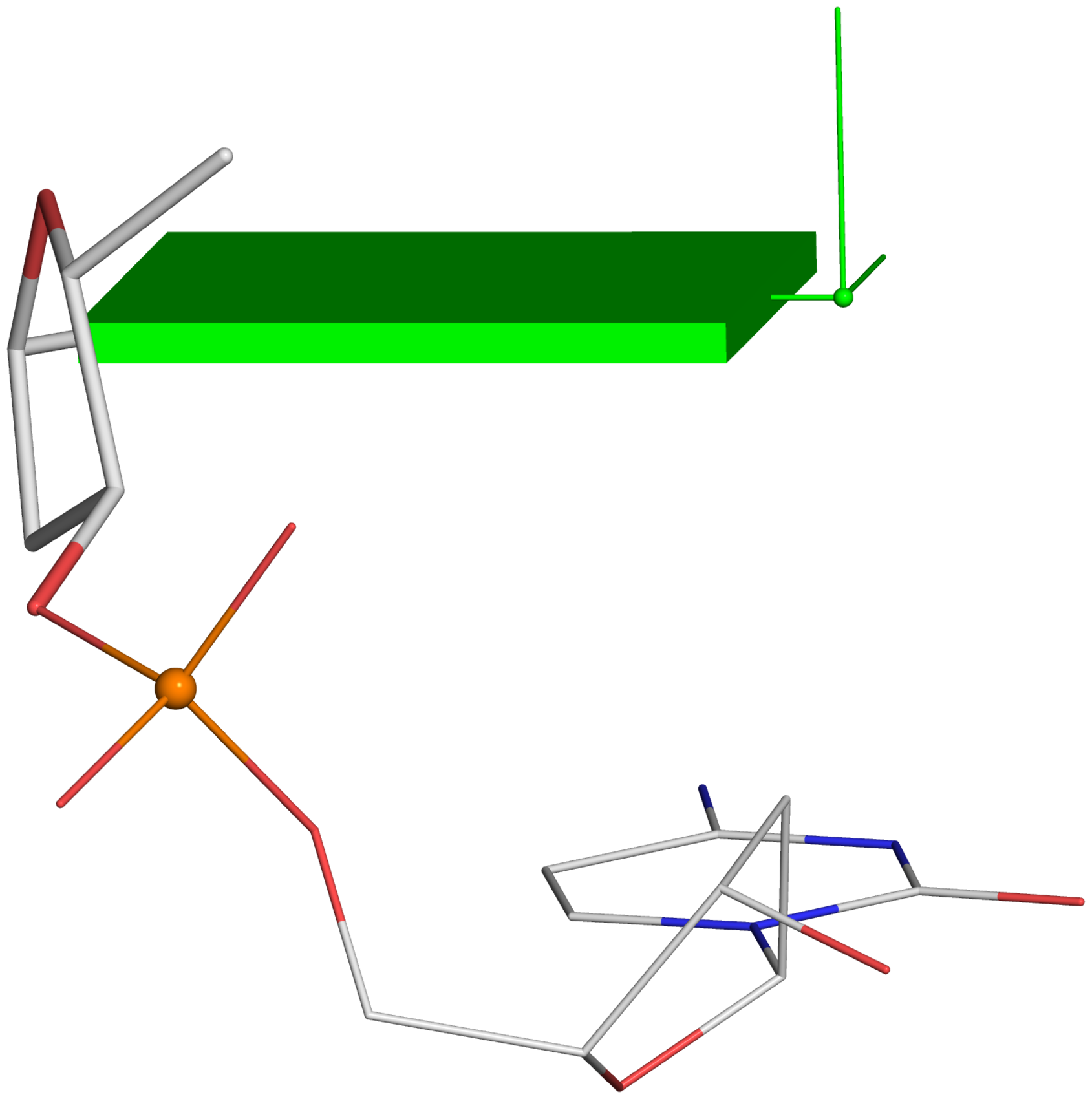 -- Fig. 2D
-- Fig. 2D





{kind=link}