This post is based on a series of email exchanges I had with Satoshi Yokojima (Institute of Materials Science, University of Tsukuba, Japan) in June 2004. This topic is on how to build base-only fiber models with varying rise, using a combination of various 3DNA components. Satoshi Yokojima kindly summarized the procedure from a user's perspective, as enclosed below.
-- Xiang-Jun
From yokojima@adenine.ims.tsukuba.ac.jp
Date: Fri, 25 Jun 2004 21:32:13 +0900 (JST)
From: Satoshi Yokojima <yokojima@adenine.ims.tsukuba.ac.jp>
To: Xiangjun LU <xiangjun@rutchem.rutgers.edu>
Cc: W. K. Olson <olson@rutchem.rutgers.edu>,
Satoshi Yokojima <yokojima@adenine.ims.tsukuba.ac.jp>
Subject: Re: 3DNA --- building "modified" fiber models
Dear Dr. Lu:
I have finished the check of the new geometry and I found that
it is what I wanted to have. The procedure you have suggested
worked nicely. The thing what I did is as follows:
[1] A new directory X3DNA/MY_Fiber55 is created.
[2] The A|C|G|T.pdb files are copied from X3DNA/FIBER/Str55
into X3DNA/MY_Fiber55.
[3] In order to rebuilt the structure without backbones,
I have deleted the backbone atoms from A|C|G|T.pdb
using vi.
[4] The following commands are executed in X3DNA/MY_Fiber55.
> std_base A.pdb Atomic_A.pdb
> std_base C.pdb Atomic_C.pdb
> std_base G.pdb Atomic_G.pdb
> std_base T.pdb Atomic_T.pdb
[5] A new directory test/rise=10 is created where I wanted to
produce the 55-th fiber model structure without the
backbone and with Rise = 10 Angstrom.
[6] The files Atomic_A|C|G|T.pdb are linked to the directory
test/rise=10.
[7] The 55-th fiber model structure 5'-GG-3' is generated in
the directory test/rise=10 by the command:
> echo "2" > f55.in
> echo "GG" >> f55.in
> echo "1" >> f55.in
> fiber -55 f55.pdb < f55.in >& f55.out
[8] The following command is executed.
> find_pair f55.pdb stdout | analyze
[9] The Rise value in the file "bp_step.par" is modified
to 10 Angstrom by vi editor.
[10] The 55-th fiber model structure without the backbone
and with Rise = 10 Angstrom is created by the command
rebuild as follows:
> rebuild -atomic bp_step.par f55_newRise.pdb
[11] The procedure from [5]-[10] is repeated for the different
Rise values.
I think it is a good idea to add a lot of examples in the manual.
People can learn much easier from the casebook than from the
explanation of the commands. I did not even imagine that the 3DNA
can do some of these things. Also, if you have a good manual,
I can recommend other people to use 3DNA to build DNA structures.
It is very kind of you to explain the detailed usage of 3DNA.
Thank you very much, again.
Sincerely yours,
Satoshi Yokojima
--------------------------------------
Satoshi Yokojima
yokojima@adenine.ims.tsukuba.ac.jp
Institute of Materials Science
University of Tsukuba
--------------------------------------
On Fri, 25 Jun 2004, Satoshi Yokojima wrote:
> Dear Dr. Lu:
>
> Thank you very much for your quick reply. It looks like it is
> working this time, but I think I need to do more careful check
> tomorrow. I will send you another e-mail when I finish doing it.
>
> Thank you very much, again.
>
> Sincerely yours,
> Satoshi Yokojima
>
> --------------------------------------
> Satoshi Yokojima
> yokojima@adenine.ims.tsukuba.ac.jp
> Institute of Materials Science
> University of Tsukuba
> --------------------------------------
>
> On Thu, 24 Jun 2004, Xiangjun LU wrote:
>
> > Dear Satoshi:
> >
> > Thanks for your message. I am so glad you noticed this slight difference.
> > Apparently, you are a rigorous scientist by not taking your tools blindly.
> >
> > The reason for this slight difference is due to the fact that the standard
> > default base geometry (Atomic_A|C|G|T.pdb under X3DNA/BASEPARS) used by
> > 3DNA when rebuilding the structure is DIFFERENT from the geometry of the
> > fiber model 55 repeating units.
> >
> > Using a different set of base geometry for analysis and rebuild is
> > expected when we developed 3DNA, and the solution to your problem would be
> > as follows:
> >
> > [1] create a new directory, say MY_Fiber55
> > [2] copy into MY_Fiber55 the A|C|G|T.pdb files from X3DNA/FIBER/Str55
> > [3] Since you would like the rebuilt structure WITHOUT backbones, you need
> > to delete the backbone atoms from A.pdb etc manually, using any text
> > editor (e.g., vi or emacs)
> > [4] run command
> > std_base A.pdb Atomic_A.pdb
> > and repeat the above for C.pdb, G.pdb and T.pdb
> > [5] Then `analyze', change Rise in "bp_step.par", and `rebuild' as
> > outlined previously, the "slight difference between the required and
> > produced structure" will be gone.
> >
> > Type "std_base" for more help information. Another issue to note here is
> > that `analyze' and `rebuild' check for standard base geometry files from
> > current working directory first, then those defined by environmental
> > variable X3DNA, and finally ~/X3DNA.
> >
> > Please have a try and let me know what happens. It would help if you could
> > document step-by-step what you did. We are currently working to improve
> > documentations on 3DNA, and we certainly welcome users working examples.
> > Of course, we will properly acknowledge your contributions.
> >
> > Best regards,
> >
> > Xiang-Jun
> >
> > On Thu, 24 Jun 2004, Satoshi Yokojima wrote:
> >
> > > Dear Dr. Lu:
> > >
> > > After examining the file produced by the method you have suggested,
> > > I have noticed that there is a slight difference between the required
> > > and produced structure. The difference is in, for example, the bond
> > > length.
> > >
> > > I wanted to keep all the structural parameters in the 55-th
> > > fiber model but Rise. It means I need to use the bond length and
> > > angles (including Hydrogen-bond) found in the 55-th fiber model.
> > > This is important because the 0.1 Angstrom difference of the bond
> > > length changes the single bond to the double bond. Therefore, the
> > > results of the electronic structure calculations are quite different.
> > >
> > > Are there any method I can use to make 3DNA produce the structure
> > > I want to have? Somewhat hard way, such as replacing BASEPAIR files,
> > > is fine for me, but I need to know what is the correct way to do it.
> > >
> > > Sincerely yours,
> > > Satoshi Yokojima
> > >
> > > --------------------------------------
> > > Satoshi Yokojima
> > > yokojima@adenine.ims.tsukuba.ac.jp
> > > Institute of Materials Science
> > > University of Tsukuba
> > > --------------------------------------
> > >
> > >
> > > On Thu, 24 Jun 2004, Satoshi Yokojima wrote:
> > >
> > > > Dear Dr. Lu:
> > > >
> > > > Thank you very much for your detailed explanation about the coordinates
> > > > used in the fiber model. Your suggested method to make the 55th fiber
> > > > model without a backbone and a different Rise value works perfectly.
> > > > That was just what I wanted to know.
> > > >
> > > > Thank you very much, again.
> > > >
> > > > Sincerely yours,
> > > > Satoshi Yokojima
> > > >
> > > > --------------------------------------
> > > > Satoshi Yokojima
> > > > yokojima@adenine.ims.tsukuba.ac.jp
> > > > Institute of Materials Science
> > > > University of Tsukuba
> > > > --------------------------------------
> > > >
> > > > On Wed, 23 Jun 2004, Xiangjun LU wrote:
> > > >
> > > > > Dear Satoshi:
> > > > >
> > > > > Thanks for using 3DNA and your nice words about it.
> > > > >
> > > > > Now, to answer your questions. Firstly, all the fiber models are based on
> > > > > literature work. Under directory X3DNA/FIBER, there is a README file, and
> > > > > a subdirectory for each of the 55 fiber models inlcuded with 3DNA. For the
> > > > > 55th fiber model, for example, you will find the following files at Str55/
> > > > >
> > > > > ----------------------------------------------------------
> > > > > A.pdb C.pdb G.pdb T.pdb
> > > > > A.rpt C.rpt G.rpt T.rpt
> > > > > TableIV.dat
> > > > > ----------------------------------------------------------
> > > > >
> > > > > File "TableIV.dat" contains the data originally published by the authors.
> > > > > In this case, S. Premilat & G. Albiser "Conformations of A-DNA and B-DNA
> > > > > in agreement with fiber X-ray and infrared dichroism." Nucleic Acids
> > > > > Research, 11(6), (1983), p.1897-1908.
> > > > >
> > > > > A|C|G|T.rpt are the processed files in "pseudo"-PDB format, actually
> > > > > storing atomic cylindrical coordinates. These repeating units are used by
> > > > > 3DNA to generate the fiber models given a base sequence or the number of
> > > > > repeats. The corresponding *.pdb files are in real PDB format with the
> > > > > corresponding x-, y-, and z-coordinates converted from the cylindrical
> > > > > coordinates. These PDB files can be displayed using a molecular graphics
> > > > > program, such as Rasmol. While they are not directly used by 3DNA while
> > > > > generating the fiber models, they were helpful for "quality-control" in
> > > > > the initial process.
> > > > >
> > > > > The 55 fiber model collected in 3DNA are from different authors, spanning
> > > > > several decades. Quite naturally, file formats, conventations for z-axis
> > > > > directions, atomic names etc, varied greatly, and of course, with typos,
> > > > > errors as well. To provide a useful tool to the community, we went through
> > > > > some quite painstaking procedures to make our collection as transparent
> > > > > and consistent as possible.
> > > > >
> > > > > Secondly, with the above background information, and if I understand your
> > > > > question accurately, i.e. you would like a model based on fiber #55,
> > > > > without the backbone and a different Rise value. This could be easily done
> > > > > in 3DNA as follows:
> > > > >
> > > > > [1] generate the 55-th fiber model using any base sequence you are
> > > > > interested in, say named f55.pdb
> > > > > [2] find_pair f55.pdb stdout | analyze
> > > > > [3] modify file "bp_step.par" or "bp_helical.par" generated by [2] using
> > > > > your favored text editor by changing the Rise values
> > > > > [4] rebuild -atomic bp_step.par f55_newRise.pdb
> > > > >
> > > > > Hope this helps.
> > > > >
> > > > > Xiang-Jun
> > > > >
> > > > > On Thu, 24 Jun 2004, Satoshi Yokojima wrote:
> > > > >
> > > > > > Dear Dr. Lu:
> > > > > >
> > > > > > I am using your program 3DNA. Thanks to your nice work,
> > > > > > it is very useful for my research.
> > > > > >
> > > > > > Recently, I wanted to make the 55-th fiber model structure
> > > > > > but without backbone and give different Rise for an analysis
> > > > > > of the energetics of DNA. Since I cannot do such a thing
> > > > > > by 3DNA as far as I understand, I tried to make a simple
> > > > > > program to do it.
> > > > > > Then, I have noticed that I do not understand the frame well.
> > > > > > At first, I thought that the i-th local base-pair frame should
> > > > > > be used as a coordinate system to find Shift, Slide, Rise
> > > > > > between the i-th and (i+1)-th base-pairs translation. However,
> > > > > > it looks like it is not the case. Now I think that the helical
> > > > > > axis is taken as the z-direction of the coordinate system but
> > > > > > that's not enough for me to determine your coordinate system.
> > > > > >
> > > > > > Could you kindly explain how you have taken the coordinate system
> > > > > > in 3DNA to calculate Shift, Slide, Rise. If it is not so easy,
> > > > > > could you tell me any reference?
> > > > > >
> > > > > > Thank you very much in advance.
> > > > > >
> > > > > > Sincerely yours,
> > > > > > Satoshi Yokojima
> > > > > >
> > > > > > --------------------------------------
> > > > > > Satoshi Yokojima
> > > > > > yokojima@adenine.ims.tsukuba.ac.jp
> > > > > > Institute of Materials Science
> > > > > > University of Tsukuba
> > > > > > --------------------------------------
> > > > > >
> > > > >
> > > >
> > >
> >
>


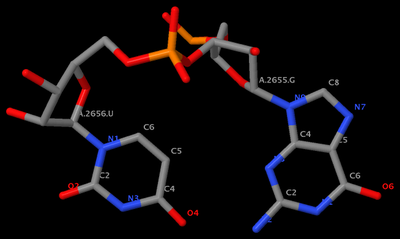
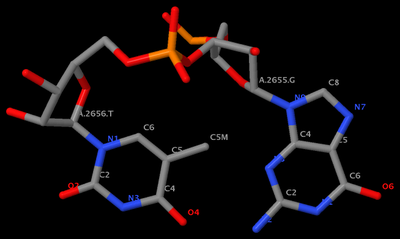
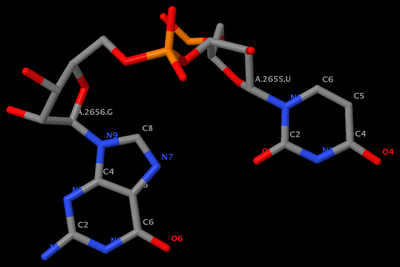
 . In the process, I've consolidated and refined the two Ruby scripts ('x3dna_md.rb' and 'extract_par.rb') for the analysis of MODEL/ENDMDL delineated molecular dynamics trajectories and NMR ensembles. For consistency, I've renamed them 'ensemble_analyze' and 'ensemble_extract'.
. In the process, I've consolidated and refined the two Ruby scripts ('x3dna_md.rb' and 'extract_par.rb') for the analysis of MODEL/ENDMDL delineated molecular dynamics trajectories and NMR ensembles. For consistency, I've renamed them 'ensemble_analyze' and 'ensemble_extract'. 